- Chi tiết
-
Được đăng: 17 Tháng 5 2017
TS. BS NGUYỄN VĂN THÀNH, TS.BS NGUYỄN THANH HỒI, ThS. BS LÊ HOÀN, ThS. BS NGUYỄN NHƯ VINH.
Với tư cách Ban soạn thảo, đại diện cho Hội thảo đồng thuận chuyên gia Hội Lao và Bệnh phổi Việt Nam được tổ chức tại Hà Nội ngày 22 tháng 4 năm 2017.
Thành viên Ban chuyên gia:
PGS.TS.BS ĐINH NGỌC SỸ (Trưởng ban), PGS.TS.BS TRẦN QUANG PHỤC, PGS. TS. BS VŨ VĂN GIÁP, GS.TS.BS ĐỖ QUYẾT, PGS. TS. BS TẠ BÁ THẮNG, PGS. TS. BS NGUYỄN VIẾT NHUNG, ThS. BS VŨ VĂN THÀNH, BS NGUYỄN MINH SANG, PGS. TS. BS TRẦN VĂN NGỌC, TS. BS LÊ THƯỢNG VŨ, ThS. BS LÊ THỊ THU HƯƠNG, BS CK II NGUYỄN ĐÌNH DUY, TS. BS ĐỖ THỊ TƯỜNG OANH, ThS. BS CAO THỊ MỸ THÚY, ThS. BS HUỲNH ANH TUẤN
Đây là tài liệu chính thức của Hội Lao và Bệnh phổi Việt Nam đã được chủ tịch phê duyệt tháng 4 năm 2017. Những khuyến cáo trong tài liệu này thể hiện quan điểm của Hội lao và bệnh phổi Việt Nam. Việc áp dụng các khuyến cáo trong tài liệu là không bắt buộc và tài liệu này không thay thế trách nhiệm của cán bộ y tế khi đưa ra quyết định trong từng hoàn cảnh cụ thể trên người bệnh và điều kiện thực tế. Các nhà quản lý y tế, lãnh đạo các cơ sở cung cấp dịch vụ chăm sóc y tế nên xây dựng quy trình thực hành của địa phương và cơ sở của mình trên nền tảng của các khuyến cáo trong tài liệu này.
Tài liệu này cũng thể hiện mong muốn sẽ có các nghiên cứu ở Việt Nam nhằm đánh giá và xác nhận giá trị nội dung và thông tin trong tài liệu này.
1. Đặt vấn đề:
COPD được định nghĩa bằng tình trạng tắc nghẽn luồng khí thở (airflow obstruction) vẫn tồn tại sau khi sử dụng thuốc dãn phế quản, tỷ lệ FEV1 (thể tích thở ra gắng sức trong một giây đầu) / FVC (dung tích sống gắng sức) sau sử dụng thuốc < 0,7. Tỷ lệ này có thể hồi phục so với trước sử dụng thuốc và theo thời gian nhưng không hoàn toàn và thông thường là tiến triển xấu dần (1). Biểu hiện lâm sàng đặc trưng là triệu chứng hô hấp dai dẳng và giảm khả năng gắng sức. Nguyên nhân thông thường là do ô nhiễm khí thở mà trong đó thuốc lá có vai trò rất quan trọng.
Tiêu chuẩn chẩn đoán bệnh hiện nay đang được áp dụng dựa trên triệu chứng lâm sàng hô hấp, yếu tố tiếp xúc ô nhiễm và sự hiện diện của tình trạng tắc nghẽn cố định luồng khí thở. Sự thiếu sót trong các tiêu chuẩn chẩn đoán này là ở chỗ nó không phản ánh được trực tiếp đặc điểm tổn thương mô bệnh học vốn rất khác nhau trong COPD, nó không tích hợp được những thay đổi phức tạp về giải phẫu bệnh học ở phổi với các thoái biến chức năng, nó không bao hàm được các kiểu viêm đường thở khác nhau, nó không định nghĩa được các kiểu biến đổi chức năng khác nhau cũng như các biến đổi cấu trúc có thể nhận thấy trên hình ảnh học (2). Việc sử dụng tỷ lệ cố định FEV1/FVC để xác định tình trạng tắc nghẽn luồng khí thở có thể đã tạo ra tình trạng chẩn đoán bệnh quá mức ở người già bình thường và dưới mức ở người trẻ có bệnh nhưng có giá trị FEV1/FVC bình thường (3,4). Mặc dù hiện nay việc điều trị COPD đã được xác định là giúp làm cải thiện triệu chứng, tăng khả năng gắng sức, giảm đợt cấp nhưng lợi ích này vẫn còn quá nhỏ và chỉ giới hạn trong một phân nhóm bệnh nhân (1).
Tình trạng viêm trên đường thở bệnh nhân COPD tỏ ra là cách đáp ứng bất thường đối với tác nhân kích thích mạn tính (thí dụ khói thuốc lá). Cơ chế làm gia tăng tình trạng viêm vẫn còn chưa được biết một cách thấu đáo. Tuy nhiên, ít nhất là một phần, cơ chế này chịu sự tác động của yếu tố cơ địa (di truyền). Bên cạnh đó, các giả thuyết về vai trò của nhiễm khuẩn quần cư (colonization), thiếu hụt anpha1-antitrypsin và sự hình thành phản ứng tự miễn dịch là cơ sở để giải thích những trường hợp COPD không hút thuốc hoặc tình trạng viêm vẫn tồn tại sau khi ngưng hút thuốc lá. Tính không đồng nhất về bản chất bệnh học đã cho thấy cách tiếp cận điều trị theo một chuẩn đơn (thí dụ theo mức độ giảm FEV1) là không hợp lý (5). Tuy nhiên, càng nhiều thông tin cần tích hợp trong phân loại bệnh nhân càng làm cho thực hành trở nên phức tạp, khó thực hiện và không tránh khỏi việc không tuân thủ điều trị theo hướng dẫn (6). Trong một bài báo gần đây (năm 2015), Claudio Tantucci và cs (7) cho rằng trị liệu cần phải hướng tới bản chất bệnh học phổ biến của COPD, độc lập với mức độ giảm FEV1 và mức độ khó thở mạn tính. Trên nền tảng như vậy, việc tập trung xác định nhóm bệnh nhân nhiều đợt cấp và điều trị nhằm làm giảm đợt cấp là một cách nhìn nhận mới về COPD.
Gần đây các tài liệu hướng dẫn (guideline) đã khuyến cáo hướng tiếp cận dựa trên các phân nhóm bệnh nhân có tiên lượng bệnh và đáp ứng điều trị giống nhau. Điều này sẽ giúp nâng cao tính hợp lý và hiệu quả điều trị. Sự hiểu biết về mối tương tác giữa gen - môi trường làm nền tảng giúp giải thích cơ chế kết hợp giữa tác nhân ô nhiễm từ môi trường và phản ứng của cơ thể chủ đưa đến phá hủy, tái tạo và nhiễm khuẩn đã hình thành chiến lược tiếp cận điều trị phù hợp hơn, chiến lược điều trị theo cá thể (personalization of therapies). Nhiều tài liệu hướng dẫn quốc gia và quốc tế đã đi theo chiến lược này. Theo chiến lược này, một mô hình tiếp cận khả thi nhất trong tình hình thực tế Việt Nam (thuốc, năng lực tiếp cận kỹ thuật y tế, cấu trúc của hệ thống chăm sóc và những tác động từ phía người bệnh) cần được xem xét và đề xuất.
2. Viêm, nhiễm trùng, tính tăng phản ứng phế quản và tự miễn dịch trong COPD
Từ nhận thức ban đầu về rối loạn chức năng thông khí kiểu tắc nghẽn đến xem bản chất viêm mạn tính là nền tảng dẫn đến các thoái biến cấu trúc, hình thành triệu chứng, có sự tác động rất khác nhau của yếu tố cơ thể chủ (host factors) (1) là một con đường dài của nhận thức về bản chất bệnh học trong COPD.
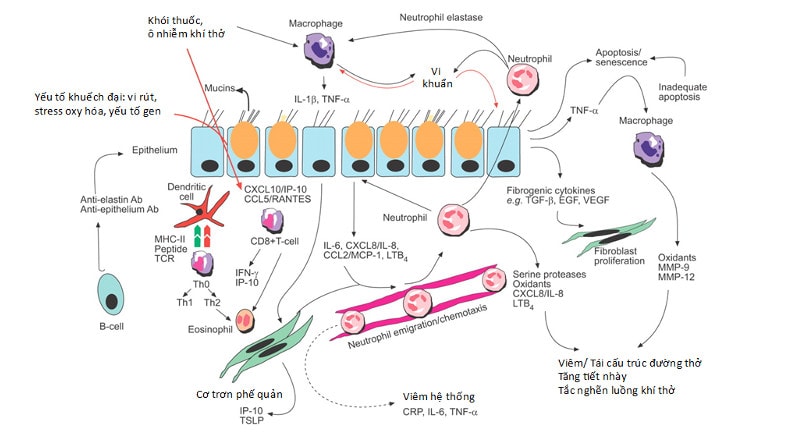
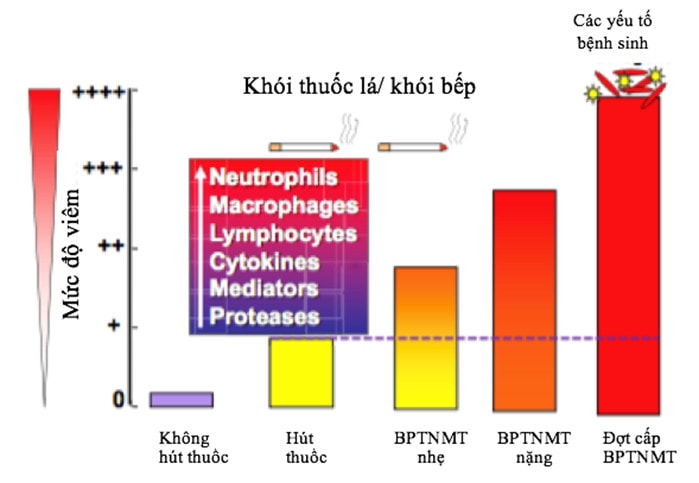
Hình 1. Sơ đồ tóm tắt phản ứng viêm và tương tác tế bào khi có tiếp xúc với thuốc lá mạn tính để hình thành tình trạng viêm mạn tính trong bệnh phổi tắc nghẽn mạn tính (COPD). Hoạt hóa các bạch cầu trung tính, đại thực bào, các tế bào biểu mô, tế bào đuôi gai, tế bào T, tế bào B, các nguyên bào sợi và tế bào cơ trơn đường thở dẫn đến giải phóng các cytokine, chemokine và protease. Khuếch đại tín hiệu là rất quan trọng trong việc làm tăng các phản ứng viêm làm nền tảng bệnh học cho COPD. Viết tắt: ab: antibody; Th: T-helper cell; MHC: major histocompatibility complex; TCR: T-cell receptor; CXCL: CXC chemokine ligand; IP: interferon (IFN)-c-inducible protein; CCL: CC chemokine ligand; RANTES: regulated on activation, normal T-cell expressed and secreted; TSLP: thymic stromal lymphopoietin; IL: interleukin; TNF: tumour necrosis factor; MCP: monocyte chemotactic protein; LT: leukotriene; CRP: C-reactive protein; TGF: transforming growth factor; EGF: epidermal growth factor; VEGF: vascular endothelial growth factor; MMP: matrix metalloproteinase (Nguồn: K.F. Chung et al. Eur Respir J 2008; 31: 1334–1356).
Viêm mạn tính trong COPD:
Viêm mạn tính trong COPD là đặc tính phản ứng của cơ thể biểu hiện bằng sự tập trung của nhiều loại tế bào, gồm neutrophils, macrophages, B-cells, nang lympho (lymphoid aggregates) và CD8+ T-cells, nhất là ở khu vực đường thở nhỏ. Cùng với hiện tượng này, sự tương tác giữa các tế bào, thành phần tế bào dưới tác động của khói thuốc, nhiễm trùng và các sản phẩm từ sự phá hủy cấu trúc đã tạo ra các cơ chế phức tạp duy trì và phát triển tình trạng viêm mà cho đến nay còn nhiều điều chúng ta chưa hiểu rõ (8) (hình 1).
Phá hủy cấu trúc ở tầm phế nang:
Bạch cầu đa nhân trung tính (BCĐNTT) hoạt hóa giải phóng ra các men tiêu hủy chất trun (elastin) của nhu mô phổi. Ở người khỏe mạnh, hoạt động này của BCĐNTT được giữ trong một trạng thái thăng bằng giữa men tiêu hủy protein và men đối kháng (proteinases/ antiproteinases). Sự mất thăng bằng đưa đến phá hủy cấu trúc phế nang được nhận thấy ở những người có thiếu hụt anpha1-antitrypsin, đặc trưng bằng hiện tượng xuất hiện khí phế thũng sớm (9). Tuy nhiên mất thăng bằng proteinases / antiproteinases có phải là cơ chế chính chịu trách nhiệm hình thành khí phế thũng hay không vẫn còn không rõ ràng vì, trong nhiều trường hợp, bệnh nhân không có biểu hiện của mất thăng bằng. Bên cạnh đó, đại thực bào cũng là tế bào có mặt nhiều hơn trên đường thở bệnh nhân COPD giống như BCĐNTT và khi hoạt hóa, tế bào này cũng giải phóng các men tiêu hủy protein (như matrix metalloproteinases, cathepsins và collagenases) làm mất cấu trúc khung gian bào của nhu mô phổi. Ở những trường hợp khí phế thũng, các nghiên cứu còn nhận thấy có hiện tượng các tế bào tăng chết theo chương trình (apoptosis) mà cơ chế có thể do giảm tiếp nhận thông tin từ yếu tố tăng trưởng nội mạc mạch máu VEGF (vascular endothelial growth factor), yếu tố giúp duy trì sự sống của tế bào (10,11). Thêm một cơ chế nữa trong sự hình thành mở rộng khoảng khí và khí phế thũng là giảm chất diện hoạt (surfactant) phổi do tế bào phế nang type II tiết ra (12).
Tái tạo (remodeling) đường thở nhỏ:
Thuật ngữ tái tạo đường thở nhỏ muốn nói đến các biến đổi tầm tế bào và cấu trúc thành đường thở. Những thay đổi theo hướng tái tạo có trong COPD bao gồm những thay đổi của cấu trúc bình thường niêm mạc biểu mô lông chuyển (epithelial cilia) và thay vào đó là hiện tượng dị sản gai (squamous metaplasia), quá sản tế bào tiết nhầy, phì đại các tuyến chế tiết dưới niêm mạc, phì đại cơ trơn phế quản, xâm nhập tế bào viêm và xơ hóa cấu trúc thành phế quản (13-15). Đường thở nhỏ là vị trí chủ yếu của cơ chế sinh bệnh tắc nghẽn trong COPD. Tắc nghẽn luồng khí thở là kết quả của cả yếu tố tái tạo, thu hẹp và tích tụ dịch viêm trong lòng đường thở. Những hiện tượng bệnh lý này tăng lên cùng với mức độ nặng của bệnh (16,17). Bên cạnh hiện tượng thu hẹp, bằng kỹ thuật vi CT (microCT) còn cho thấy có sự biến mất của một số tiểu phế quản tận, góp phần làm gia tăng trở kháng đường thở ngoại vi trong COPD (18).
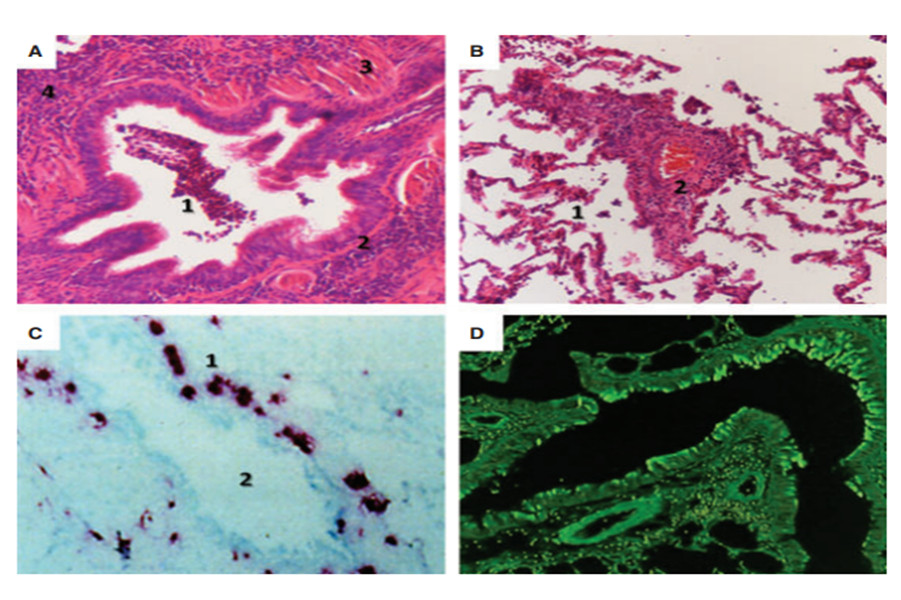
Sự tồn tại mạn tính của vi khuẩn trên đường thở:
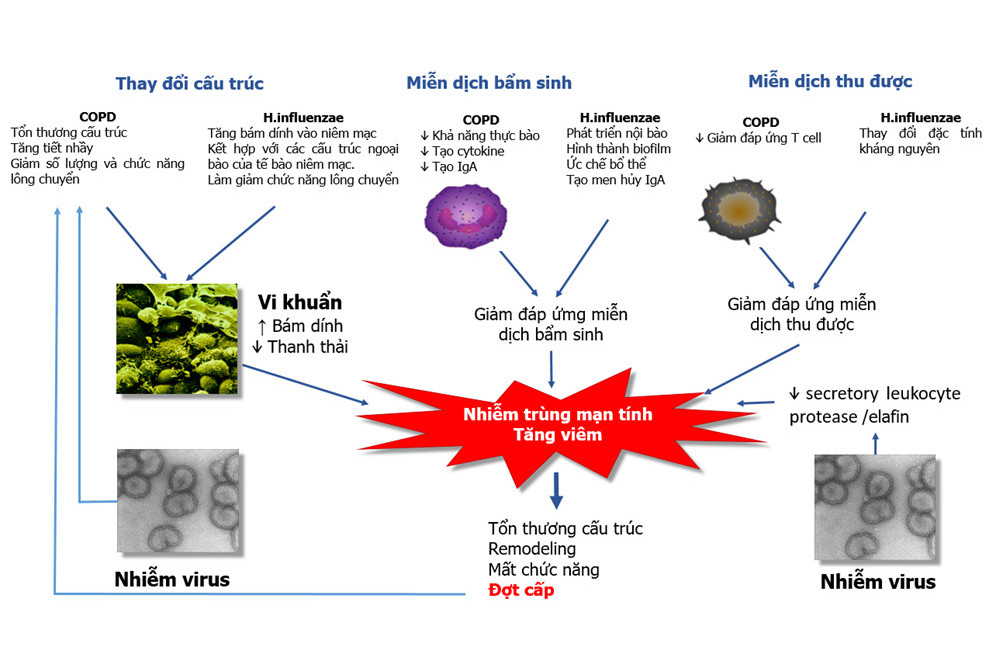
Với chức năng là một cơ quan mở, đường thở luôn có sẵn những cơ chế bảo vệ bẩm sinh hay thu được nhằm bảo vệ sự toàn vẹn về hình thái và chức năng trước các tác động thường xuyên của rất nhiều yếu tố từ môi trường. Khói thuốc lá làm giảm năng lực hoạt động chức năng của hệ thống nhầy-lông chuyển (19), tạo điều kiện thuận lợi cho vi khuẩn xâm nhập và bám dính vào được bề mặt niêm mạc (20), đồng thời cũng làm giảm chức năng bảo vệ của niêm mạc (21,22), trong đó có chức năng thực bào của các đại thực bào (23). Những bất thường của hoạt động bảo vệ này vẫn còn tồn tại ngay cả khi đã ngưng hút thuốc lá (24). Ở bệnh nhân COPD, mặc dù lượng đại thực bào trong đường thở tăng nhưng khả năng thực bào của chúng lại giảm. Đã có bằng chứng trên người hút thuốc lá, khả năng thực bào của đại thực bào giảm đối với H.influenzae và S.pneumoniae so với người không hút thuốc lá (25,26). Khả năng thực bào giảm cùng với các suy giảm khác của chức năng nhầy lông chuyển tạo điều kiện thuận lợi cho vi khuẩn hình thành màng bám sinh học (biofilm) và vi khuẩn quần cư (colonization). Trên nền tảng này, các kích thích từ nhiễm trùng sẽ góp phần duy trì phản ứng viêm mạn tính trên đường thở (hình 2). Cũng với nền tảng suy giảm năng lực miễn dịch, bệnh nhân COPD có khả năng dễ nhiễm virus có thể do giảm tiết interferon. Mặt khác, nhiễm virus cũng là điều kiện thuận lợi cho nhiễm khuẩn. Sự xuất hiện nhiều hơn tế bào lympho T CD8(+) ở đường thở nhỏ có thể là hậu quả của nhiễm virus tái diễn và colonization (27).
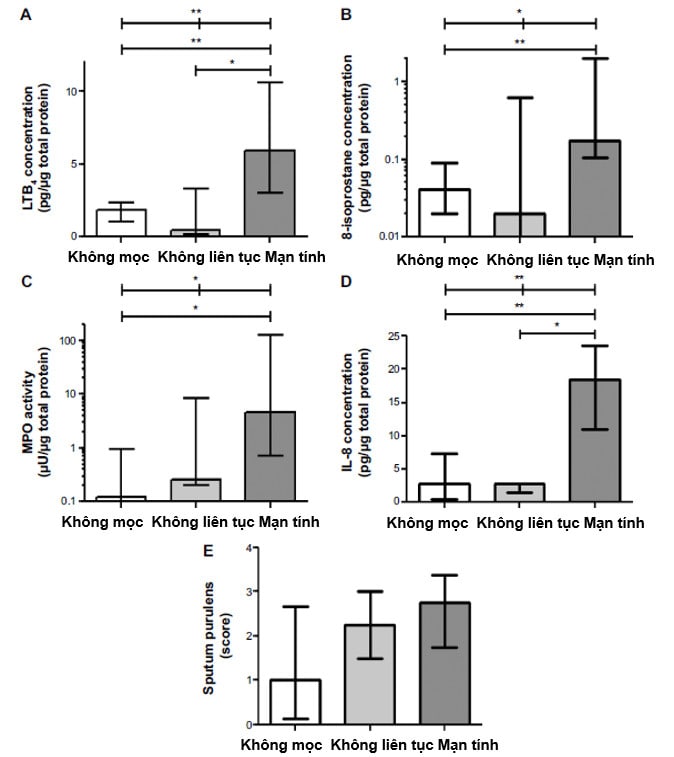
Hình 2. Các biomarker đàm (LTB4 (A), 8-isoprostane (B), MPO activity (C), IL-8 concentration (D), Điểm đàm đục (E) trên bệnh nhân không có colonization H.influenzae, có nhưng không liên tục và có mạn tính. Dữ liệu thể hiện trị số trung bình. (*): p<0,05; (**): p<0,01. Viết tắt: LTB4, leukotriene B4; MPO, myeloperoxidase; IL, interleukin (Nguồn: Ellen Tufvesson và cs. International Journal of COPD 2015:10 881–889)
Tương tác bệnh lý giữa yếu tố cơ thể chủ và tác động của môi trường:
Như vậy trên người COPD, ngay cả khi không hút thuốc lá hay đã ngưng hút thuốc lá, vẫn có hai nguồn kích thích viêm thường trực: các sản phẩm từ sự phá hủy cấu trúc và vi khuẩn tồn tại mạn tính. Trên cơ sở này, hai cơ chế bảo vệ bằng hình thức đáp ứng phân tử sẽ được khuếch đại: cơ chế đáp ứng với các sản phẩm phá hủy, DAMP (damage-associated molecular pattern) và cơ chế đáp ứng với nhiễm khuẩn, PAMP (pathogen-associated molecular pattern)(27). Giảm IgA chế tiết đã được chứng minh là có kết hợp với sự xâm nhập của vi khuẩn vào niêm mạc đường thở và các biến đổi cấu trúc mạn tính (remodeling) (28). Có sự liên quan rất chặt giữa giảm IgAs trên niêm mạc đường thở nhỏ, nhiễm virus tiềm tàng, tăng lympho T CD8 với biến đổi cấu trúc và tình trạng tắc nghẽn đường thở (28). Trong một bài tổng quan, M.G. Cosio Piqueras và cs (năm 2001) (29) nhận định trên người hút thuốc lá, sự hiện diện của T CD8+ tạo ra sự khác biệt giữa người hút thuốc lá COPD và người hút thuốc lá không COPD. Bên cạnh đó, các tác giả cũng cho rằng với cùng một mức độ tắc nghẽn, mức độ viêm và các biến đổi ở đường thở tùy thuộc nhiều vào kiểu hình thành khí phế thũng trung tâm tiểu thùy hay toàn bộ tiểu thùy. Khí phế thũng dạng trung tâm tiểu thùy có biểu hiện biến đổi viêm và thu hẹp lòng đường thở nặng hơn. Như vậy có thể thấy rằng trước những tác động có vẻ như nhau phản ứng xảy ra khác nhau giữa người COPD với người không COPD, khác nhau giữa người COPD này với người COPD khác. Nền tảng của sự khác nhau này là đặc tính viêm và các biến đổi cấu trúc.
Tính tăng phản ứng đường thở trong COPD:
Năm 1961, “giả thuyết Hà Lan” (Dutch hypothesis) lần đầu tiên được Orie và nhóm nghiên cứu công bố. Giả thuyết này cho rằng cả yếu tố di truyền (gen) và yếu tố môi trường quyết định ai là người sẽ bị bệnh đường thở tắc nghẽn và cũng quyết định kiểu hình của bệnh lý này nếu bị bệnh. Cho đến gần đây, cùng với rất nhiều tiến bộ trong kỹ thuật xác định bản chất bệnh học COPD (như kỹ thuật sinh học phân tử, di truyền học, CT scans...), giả thuyết này lại được nhắc đến với nhận định rằng tính thời sự của giả thuyết Hà Lan vẫn còn rất quan trọng. Theo Dirkje S. Postma và cs (năm 2015) (30), khi xem lại giả thuyết Dutch, đã nhấn mạnh 3 vấn đề có liên quan tới việc quản lý và điều trị COPD, đó là: tính tăng phản ứng của đường thở (hyperresponsiveness), viêm tăng BCAT và nhiễm trùng.
Tình trạng tăng phản ứng đường thở (airway hyperresponsiveness, AHR) được định nghĩa là tính thu hẹp lòng đường thở quá dễ hoặc quá nhiều khi có kích thích. Định nghĩa này đã được áp dụng trong hen và rất nhiều bằng chứng cho rằng AHR là hình ảnh bệnh học cốt lõi trong hen. Tuy nhiên AHR còn ít được nghiên cứu và hiểu trong COPD. Trên bệnh nhân COPD, AHR có thể biểu hiện ở trên 60-90% (31-33). Có nhiều bằng chứng cho rằng AHR trong COPD không chỉ đơn thuần phản ánh tình trạng thu hẹp lòng đường thở mà còn thể hiện các bất thường về sinh bệnh học, tạo nên một dạng kiểu hình, ít nhất là ở một số bệnh nhân(34). Trong một phân tích tổng quan, N Scichilone (năm 2006) cho rằng AHR nên được xem xét như là một thành tố quan trọng trong COPD (35). Gần đây, năm 2012, Van den Berge M và cs cho rằng mức độ nặng của AHR trên bệnh nhân COPD kết hợp độc lập với cả các marker đường thở nhỏ (tỷ lệ thể tích cặn/ dung tích phổi toàn bộ) và với cả viêm đường thở (BCĐNTT, lymphocyte và đại thực bào) (32). Như vậy AHR là đặc điểm cung cấp các thông tin kiểu hình và hoạt tính của bệnh. Trong bệnh cảnh COPD, AHR được chứng minh là yếu tố dự đoán tử vong nên chiến lược giảm AHR có thể làm giảm diễn biến tự nhiên của COPD và có thể có lợi từ việc lựa chọn các trị liệu thích hợp (35).
Tăng BCAT:
Viêm tăng BCAT đường thở là biểu hiện trong đa số các trường hợp hen phế quản, đặc biệt là trên những trường hợp không được điều trị bằng corticosteroid (36). Mặc dù mức độ viêm tăng BCAT có kết hợp với triệu chứng, đợt cấp, cũng như với mức độ nặng của tắc nghẽn và AHR nhưng thường là khá thấp (37,38). Điều này gợi ý rằng viêm tăng BCAT mạn tính có thể góp phần trong việc hình thành tái tạo đường thở và phát triển tình trạng tắc nghẽn khi không được điều trị kéo dài (39). Có khoảng 40% bệnh nhân COPD, nhất là những trường hợp có tắc nghẽn đường thở nặng, có biểu hiện tăng tỷ lệ BCAT trong đàm (40-43). Năm 2011, Liesker và cs (44) chứng minh rằng bệnh nhân COPD có BCAT máu tăng >3% sẽ tăng nguy cơ đợt cấp nếu không điều trị ICS so với nhóm không tăng BCAT. Tăng BCAT không phải là hiện tượng chỉ thấy trên bệnh nhân hen và hen-COPD chồng lấp mà còn là marker viêm trong đàm quan trọng trong COPD. Khi phân tích tổ hợp (cluster analysis) các chất trung gian viêm trong đàm trên bệnh nhân hen nặng và COPD trung bình-nặng, Ghebre và cs (năm 2015) (45) xác định đặc tính viêm có sự khác biệt và cũng có cả sự chồng lấp trong hen, COPD. Trong hen có sự tăng ưu thế của BCAT và các trung gian viêm thông qua Th2, trong khi COPD tăng ưu thế các cytokine tiền viêm. Nhóm chồng lấp hen và COPD có biểu hiện lâm sàng viêm phế quản mạn tính, tăng colonization, tăng IL-beta1 và TNF-anpha cũng như tăng BCĐNTT trong đàm. Nhiều nghiên cứu cho rằng tăng BCAT trên bệnh nhân COPD kết hợp với tăng đáp ứng với trị liệu corticosteroid (46-52). Trong một nghiên cứu ngẫu nhiên có đối chứng (năm 2007), R. Siva và cs nhận thấy tăng BCAT được điều trị hướng tới làm giảm BCAT trong đàm so với điều trị thường quy theo BTS guideline sẽ làm giảm đợt cấp nặng (p=0.037), nhất là trong nhóm có tăng BCAT trong máu >3% (53). Trong một nghiên cứu khác, cũng thiết kế ngẫu nhiên có đối chứng so sánh hai nhóm đợt cấp COPD tăng và không tăng BCAT (>2% BCAT trong xét nghiệm máu), nhóm chứng được điều trị pretnisolone 2 tuần kèm kháng sinh và nhóm nghiên cứu điều trị kháng sinh có hay không pretnisolone theo hướng dẫn BCAT máu cho thấy điều trị theo hướng dẫn BCAT cho kết quả không thấp hơn điều trị thường quy cả về lâm sàng (chỉ số CRQ) và tỷ lệ thất bại (54).
Viêm và tự miễn:
Thuốc lá hoạt hóa các tế bào có chức năng miễn dịch bẩm sinh (như các tế bào niêm mạc, đại thực bào) một cách trực tiếp hay gián thông qua thông tin phân tử từ các tế bào chịu kích thích quá mức (stressed) hay tế bào chết. Các tế bào đuôi gai (hoạt hóa) có chức năng trình diện kháng nguyên dẫn đến đáp ứng miễn dịch thu được thông qua các tế bào T helper (Th1 và Th17), tế bào CD4 + T, CD8 + độc tế bào, và phản ứng tế bào B, dẫn đến sự phát triển các nang bạch huyết đáp ứng viêm mạn tính. Nhiễm virus và vi khuẩn không chỉ là căn nguyên gây ra đợt cấp mà còn tham gia làm tăng thêm, duy trì tình trạng viêm mạn tính trong COPD ổn định thông qua các cơ chế duy trì viêm mạn tính kể trên.
Vai trò của bệnh học tự miễn dịch trong sự hình thành và tiến triển của bệnh phổi tắc nghẽn mạn tính đang ngày càng được xác định. Giả thuyết về cơ chế bệnh sinh bệnh tự miễn là hướng giải thích các câu hỏi: tại sao có người hút thuốc lá bị COPD và có người không bị, tại sao tình trạng viêm vẫn duy trì sau khi đã ngưng hút thuốc lá và cơ chế hình thành đợt cấp COPD là gì. Trên cơ sở có sự hiện diện của cấu trúc lympho B ở bệnh nhân COPD nặng và sự phát hiện ra nhiều tự kháng thể trong huyết thanh ở một nhóm bệnh nhân COPD, COPD đã được xem là bệnh tự miễn (55). Nhiều nghiên cứu đã phát hiện có tự kháng thể kháng cấu trúc trun (antielastin) và đáp ứng Th1, liên quan tới mức độ nặng ở bệnh nhân COPD (56-58). Các kháng thể chống lại tế bào biểu mô nguyên phát đã được ghi nhận ở bệnh nhân COPD nhiều hơn nhóm chứng và kháng thể kháng cơ trơn ở 1/5 số bệnh nhân có tiền sử nhập viện vì đợt cấp và có liên quan tới mức độ nặng của tình trạng tắc nghẽn (56,57). Tuy nhiên mối quan hệ nhân- quả trong sinh bệnh học giữa tự miễn và cơ chế hình thành COPD vẫn còn chưa được xác lập (55).
3. Khí phế thũng và Bệnh đường thở nhỏ:
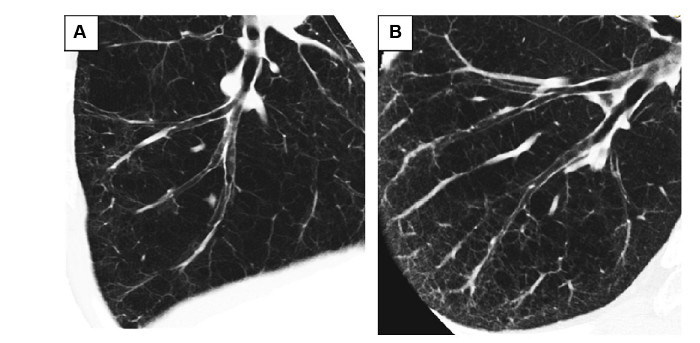
Hình 3. Hai dạng khí phế thũng. Bệnh nhân A (trái) có cấu trúc nhu mô quanh phế quản ít hơn bệnh nhân B (phải). Bệnh nhân A có chức năng phổi kém hơn và tốc độ giảm chức năng phổi nhanh hơn trong khi có cùng giá trị DLCO. Hình cắt dọc phế quản B9 phải ra ngoại vi (Nguồn: Kazuyoshi Kurashima và cs. International Journal of COPD 2015:10 1027–1033)
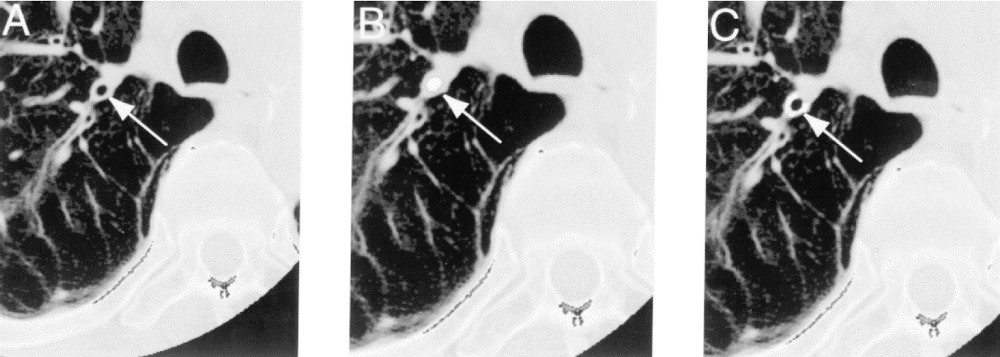
Hình 4. Phân tích đường thở bằng sử dụng một hình ảnh CT xoắn ốc đi qua phế quản phân thùy đỉnh thùy trên (mũi tên A). Sử dụng thuật toán xác định diện tích lòng (mũi tên B) và độ dày thành (mũi tên C). Lưu ý việc xác định độ dày thành đường thở được thực hiện thành công ngay cả khi các mạch máu phổi chạy song song với các phế quản (Nguồn: Nakano et al. Am J Respir Crit Care Med Vol 162. pp 1102–1108, 2000).
Trên bệnh nhân hút thuốc lá ở cùng một mức độ tắc nghẽn, các biến đổi của nhu mô xảy ra theo hai dạng hoàn toàn khác nhau: khí phế thũng ưu thế trung tâm tiểu thùy và khí phế thũng toàn bộ tiểu thùy. Hai dạng này có thể biểu hiện khá đơn độc hoặc chồng lấp với ưu thế rõ ràng của từng dạng. Mức độ bất thường của đường thở và đặc tính cơ học của nhu mô phổi thay đổi phụ thuộc vào dạng tổn thương này (59). Bằng phân tích Xquang thường quy và CT trong COPD, các nghiên cứu cho rằng bệnh nhân COPD nếu không có biểu hiện khí phế thũng trên Xquang ngực có thể sẽ là dạng COPD có tổn thương chỉ đơn thuần (exclusively) là đường thở (60).
Tình trạng tắc nghẽn cố định đặc trưng của COPD là kết quả của hai hình thái tổn thương giải phẫu bệnh: khí phế thũng và các biến đổi trên đường thở nhỏ. Khi các biến đổi tập trung chủ yếu ở nhu mô phổi, hình ảnh bệnh học là phá hủy cấu trúc nhu mô phổi và tình trạng tắc nghẽn được tạo nên do giảm độ co hồi trun (elastic recoil) của cấu trúc mô phổi và thu hẹp lòng đường thở nhỏ do giảm lực kéo ly tâm từ cấu trúc phế nang kết nối với chúng. Ngược lại, khi các biến đổi tập trung ở đường thở nhỏ sẽ tạo ra tình trạng tắc nghẽn do thu hẹp, bít tắc, biến dạng lòng đường thở (hình 3,4). Trong trường hợp này, trở kháng đường thở tăng lên do nhiều cơ chế (như thu hẹp lòng, dầy thành, tăng dịch tiết). Khi các biến đổi giải phẫu đã hình thành, có một mối tương quan chặt chẽ giữa tiến triển suy sụp chức năng hô hấp và mức độ tổn thương ở đường thở nhỏ. Ở bệnh nhân COPD nặng, tình trạng gia tăng đáp ứng viêm ở đường thở ngoại vi đã được chứng minh với tăng gấp 3 lần số lượng bạch cầu, nhất là lymphocytes và macrophages cho thấy tình trạng viêm khởi đầu do khói thuốc lá diễn biến xấu đi cùng thoái giảm chức năng hô hấp (tắc nghẽn, căng phồng phổi quá mức, giảm khuếch tán CO) và khí phế thũng (61,62). Điều này cho thấy tình trạng viêm của đường thở nhỏ đóng vai trò quan trọng trong các thoái biến chức năng có trong COPD ngay cả trên những trường hợp khí phế thũng.
Trên bệnh nhân COPD, tình trạng gia tăng đáp ứng viêm cũng được nhận thấy ở nhu mô phổi bệnh nhân khí phế thũng nặng (63). Điều đáng lưu ý ở cùng một mức độ tắc nghẽn, người hút thuốc lá có ít tổn thương khí phế thũng hơn sẽ có tổn thương phế quản nhỏ nhiều hơn và ngược lại (63). Trong một nghiên cứu hình ảnh bằng microCT, John E. McDonough và cs (năm 2011) (64) còn nhận thấy bên cạnh các biến đổi trên đường thở nhỏ như mô tả còn có hiện tượng biến mất của trên 70% số lượng hình ảnh cắt ngang tiểu phế quản và giảm trên 80% diện tích cắt ngang của các tiểu phế quản tận. Đây cũng là nguyên nhân quan trọng làm tăng trở kháng đường thở trong COPD. Những nhận định về tổn thương đường thở nhỏ cho thấy tính không đồng nhất về cách phản ứng của cơ thể đối với tác động của thuốc lá và hiển nhiên đặc tính tổn thương bệnh học cũng sẽ có tác động tới biểu hiện lâm sàng và đáp ứng điều trị.
4. Vấn đề đợt cấp trong sinh bệnh học COPD và xác định yếu tố dự đoán:
Thuật ngữ bệnh nhân COPD nhiều đợt cấp (frequent exacerbator) lần đầu xuất hiện năm 1998 trong nghiên cứu của Seemungal TA và cs (65). Điểm cắt số đợt cấp trung bình là 3 đợt cấp/năm (≤2 hoặc ≥3) cũng được đề xuất từ nghiên cứu này. Seemungal TA và cs nhận định nhiều đợt cấp biểu hiện như là một kiểu hình với tiên lượng xấu hơn (65).
Mặc dù đợt cấp COPD sẽ trở nên thường xuyên hơn và nặng hơn khi COPD tiến triển tới giai đoạn nặng nhưng có vẻ như tính tăng đợt cấp là một biểu hiện kiểu hình trên một nhóm bệnh nhân. Trong một nghiên cứu mô tả theo dõi dọc trong 3 năm (nghiên cứu ECLIPSE) đã ghi nhận một số nhận xét quan trọng về tần suất đợt cấp (66). Đợt cấp tăng theo mức độ nặng của phân loại GOLD. Số bệnh nhân có từ 2 đợt cấp trở lên theo dõi trong năm đầu 22% ở giai đoạn II, 33% ở giai đoạn III và 47% ở giai đoạn IV. Yếu tố dự đoán đợt cấp tốt nhất ở tất cả các giai đoạn là tiền sử đợt cấp. Qua theo dõi trong 3 năm, kiểu hình nhiều đợt cấp tỏ ra là khá ổn định và có thể dự đoán được trên cơ sở khả năng nhớ của người bệnh. Trên cơ sở của kết quả nghiên cứu này, các tác giả cho rằng một chiến lược hướng tới phòng đợt cấp nên thực hiện trên tất cả bệnh nhân COPD ở các giai đoạn.
Trị liệu kết hợp ICS/LABA đã được khuyến cáo trên bệnh nhân COPD nhiều đợt cấp và nặng. Tuy nhiên lợi ích của ICS kết hợp luôn được tranh luận do vấn đề về hiệu quả, tính an toàn và chi phí, nhất là khi cần điều trị kéo dài. Trong 1 nghiên cứu gần đây, nghiên cứu WISDOM (năm 2014) (67) đã kết luận trên bệnh nhân COPD nặng đang sử dụng tiotropium kết hợp salmeterol, nguy cơ có các đợt cấp trung bình, nặng là như nhau trên nhóm điều trị ICS liên tục và trên nhóm giảm dần và ngưng ICS. Tuy nhiên ở thời điểm 18 và 52 tuần, nhóm ngưng ICS có tốc độ giảm FEV1 nhanh hơn nhóm tiếp tục sử dụng ICS. Cũng cần lưu ý rằng nghiên cứu cũng không phân tích dưới nhóm và chỉ ra được có phân nhóm bệnh nhân nào có lợi từ việc sử dụng ICS trong việc làm giảm đợt cấp hay không. Cung cấp thêm thông tin để có thể lý giải hiện tượng giảm chức năng khi ngưng trị liệu ICS này bằng dữ liệu từ một nghiên cứu gần đây (năm 2017) (68) đã cho thấy có hiện tượng tăng tế bào viêm (trên bệnh phẩm sinh thiết, đàm) trên bệnh nhân COPD trung bình-nặng sau khi ngưng sử dụng ICS một thời gian dài và theo dõi. Như vậy chúng ta thêm một lần nữa phải chú ý rằng mối quan hệ giữa tăng viêm và tăng thoái giảm chức năng hô hấp có thể nằm chung trong bệnh cảnh nhiều đợt cấp và cũng có thể không.
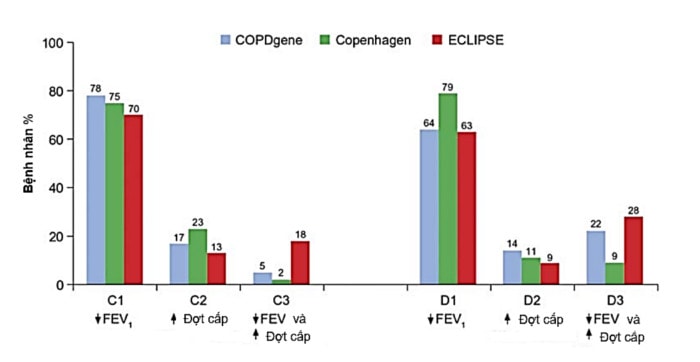
Hình 5. Phân bố tỷ lệ bệnh nhân theo GOLD nhóm C và D. Theo FEV1 < 50% giá trị ước tính: C1, D1; Theo tiền sử đợt cấp: C2, D2; Và theo cả 2: C3, D3 (số liệu từ 3 nghiên cứu lớn là COPD gene, Copenhagen và ECLIPSE). Số liệu cho thấy hầu hết bệnh nhân vào nhóm C-D là do giảm FEV1 đơn thuần (Nguồn: A. D’Urzo et al. Expert Opin. Pharmacother. 2015. 16 (12):1845-1860).
Chiến lược GOLD thiết kế vẫn hướng tới nguy cơ hơn là hướng tới kiểu hình. Chúng ta thấy rằng ICS/LABA được khuyến cáo sử dụng như là lựa chọn đầu tiên trên nhóm C và D, nhóm nguy cơ cao đợt cấp và/hoặc tắc nghẽn nặng. Với cách phân loại này, hầu hết bệnh nhân vào nhóm C và D với lý do duy nhất là tắc nghẽn (70-78% ở nhóm C và 63-79% ở nhóm D) trong khi chỉ có một tỷ lệ thấp bệnh nhân có nhiều đợt cấp (nhiều đợt cấp 9-23% hoặc vừa nhiều đợt cấp vừa giảm FEV1 2-28%) (hình 5) (69). Về đặc tính bệnh nhân được xếp vào các nhóm theo phân loại A-D, một nghiên cứu thực hành đa khoa ở Anh (năm 2014) (70) cũng cho kết quả tương tự, 70% trên 2.282 bệnh nhân có tắc nghẽn nặng có nguy cơ có đợt cấp thấp (≤1). Nhóm nặng và nhiều đợt cấp nhưng có kèm dãn phế quản tỏ ra là ít hiệu quả với điều trị ICS. Nhóm có dãn phế quản đồng thời, có thể chiếm tới 50% bệnh nhân COPD nặng, có nhiều đợt cấp, đợt cấp nhập viện và colonization (71,72).
Khái niệm kể lại mang tính chủ quan “nhiều đợt cấp” do vậy cần được xác định một cách khách quan hơn, trong đó hướng tới xác định bằng biomarker viêm toàn thân là một cách tiếp cận. Một số nghiên cứu đã xác định mối quan hệ giữa các marker viêm và tần suất đợt cấp. Có 8 biomarker viêm toàn thân được xem có khác biệt ý nghĩa giữa người hút thuốc COPD và người hút thuốc không COPD, trong đó có CRP (73). Một phân tích dưới nhóm trên 1.755 bệnh nhân COPD cho thấy có một nhóm bệnh nhân (16% trên tổng số) có ít nhất 2 trong 4 marker viêm (gồm tăng bạch cầu, tăng CRP, tăng IL-6 và tăng fibrinogen trên mức trung vị 1/4) tồn tại sau 1 năm theo dõi gắn liền với tăng đợt cấp cũng như tăng tử vong do các nguyên nhân có ý nghĩa (74). Một nghiên cứu khác (75) phân tích cohort trên 6.574 bệnh nhân COPD kết luận tăng đồng thời 3 marker kèm theo tăng đợt cấp: CRP (>3 mg/l), fibrinogen (>14 µmol/l), và bạch cầu máu (>9 x 109/l). Hơn nữa các marker viêm này có thể xác định những bệnh nhân tăng nguy cơ đợt cấp ngay cả trên những trường hợp tiền sử không có đợt cấp và nhiều đợt cấp làm tăng các marker này theo thời gian.
Thực hành lâm sàng COPD thông thường là ở y tế đa khoa và chăm sóc ban đầu. Việc xác định phenotype nhiều đợt cấp sớm là rất quan trọng. Trong 1 nghiên cứu với định hướng tìm marker có tính thực hành lâm sàng cao: có kết quả nhanh, rẻ, dễ nhận định kết quả, Aida M. Yousef (năm 2016) (76) cho rằng bên cạnh CRP, bạch cầu máu, tỷ lệ BCĐNTT/Lymphocyte cũng là một marker viêm quan trọng trong COPD. Tuy nhiên, không có 1 marker nào có thể phản ánh được đầy đủ bản chất đa hệ thống (multiscale) của phenotype COPD mà trong đó thể hiện mối tương tác gen-tế bào, tế bào-mô, mô-tạng, tạng-toàn thân (77). Trong một bài tổng quan gần đây, Brendan J. Carolan và cs (năm 2013) (78) cho rằng phenotype COPD có thể xác định dựa trên đặc điểm lâm sàng, chức năng, phân tử và hình ảnh học. Việc đánh giá cẩn thận COPD sẽ giúp cho xây dựng kế hoạch tiếp cận điều trị phù hợp với từng người bệnh. Tuy nhiên, cũng còn những điều chúng ta chưa biết như phương pháp lâm sàng nào thích hợp nhất để xác định phenotype, tính ổn định của phynotype theo thời gian, hiệu quả thực của cách tiếp cận điều trị theo phenotype.
5. Những vấn đề có tính thực hành cao hiện nay:
Định nghĩa và phân loại:
COPD với tiếng Anh là “D” (disease - bệnh) và Bệnh phổi tắc nghẽn mạn tính trong tiếng Việt đều nhìn nhận COPD là một bệnh. Với những phân tích như ở trên, COPD cần được xem là biểu hiện rối loạn chức năng (Disorder) của ít nhất hai dạng bệnh lý: nhu mô phổi và đường thở nhỏ trên nền tảng của tình trạng đáp ứng miễn dịch và viêm mạn tính, biểu hiện khác nhau giữa cá thể này với cá thể khác. Giảm chức năng thông khí, giảm giá trị tỷ lệ FEV1/FVC chỉ là biểu hiện của tình trạng rối loạn (disorder) mà không phải là bệnh. Với nhận định như vậy, khi tiếp cận bệnh nhân COPD, điều đầu tiên và quan trọng nhất là xác định bệnh lý nền tảng. Thông thường tiếp cận lâm sàng, Xquang ngực và chức năng phổi có thể đủ để xác định (79). Bên cạnh các trị liệu khác, sử dụng kéo dài các thuốc kháng viêm steroid là sự khác biệt quan trọng nhất mà trị liệu thuốc cần xác định cho từng phân nhóm bệnh. Trong một bài viết gần đây, Claudio Tantucci và cs (năm 2015) (79) đề xuất cách tiếp cận điều trị theo đặc điểm tổn thương nền tảng, chia thành ba nhóm: viêm tiểu phế quản mạn tính, viêm tiểu phế quản mạn tính kết hợp khí phế thũng dạng trung tâm tiểu thùy và khí phế thũng dạng toàn bộ tiểu thùy. Năm 2014, Tây Ban Nha đề xuất guideline (còn gọi là GesEPOC) (80) hướng tới tiếp cận theo tính chất đợt cấp và theo nhóm tổn thương ưu thế khí phế thũng hay viêm phế quản mạn tính (có thể được hiểu như là nhóm không khí phế thũng). Tuy nhiên, GesEPOC vẫn còn để một phân nhóm trung gian giữa ít và nhiều đợt cấp, nhóm hen và COPD chồng lấp (ACOS) (hình 6). ACOS, nên xem là một kiểu hình và cần được định nghĩa rõ hơn một thuật ngữ chỉ mang tính khái niệm chồng lấp lâm sàng giữa Hen-COPD như GINA nhận định (81). Thêm một khái niệm phân loại như đối với GesEPOC đề xuất là tăng thêm khó khăn cho thực hành, nhất là khi khó có điều kiện để làm cho rõ ràng bản chất bệnh học của ACOS.
Nhìn nhận ACOS từ góc độ thực hành:
Các tiêu chuẩn hướng tới chẩn đoán hen trên bệnh nhân có tình trạng tắc nghẽn cố định
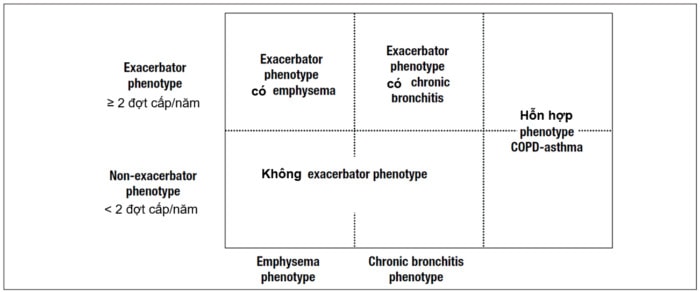
Hình 6. Phân loại phenotype lâm sàng theo GesEPOC (80)
(FEV1/FVC<0,7 sau sử dụng thuốc dãn phế quản) như: hồi phục phế quản dương tính, tiền sử hen, tiền sử atopy, tăng BCAT... đều không nhậy, không đặc hiệu và là những tiêu chuẩn có thể thấy ở một phần lớn bệnh nhân COPD (82). Hen và COPD có cùng bản chất bệnh học: viêm đường thở mạn tính, tắc nghẽn và tăng phản ứng. Hen không tăng BCAT, tăng BCĐNTT và không đáp ứng với điều trị corticosteroid đã được thông báo (83,84), và ngược lại, COPD tăng BCAT với cải thiện tốt tình trạng tắc nghẽn khi điều trị corticosteroid cũng đã được ghi nhận (85). Về mô bệnh học, cả trong hen và COPD đều thể hiện là bệnh lý của đường thở lớn (>2mm), đường thở nhỏ (<2mm) và nhu mô phổi (16,60,86). Trong tình trạng tắc nghẽn cố định (COPD và ACOS) trên CT đều có bất thường dạng biến đổi và tái tạo ở phế quản nhỏ (87,88). ACOS có nhiều đợt cấp gấp 3 lần hơn COPD đơn thuần và cũng có nguy cơ cao vào đợt cấp 2,11 lần và đợt cấp nhập viện 4,11 lần hơn COPD đơn thuần (87). Mặc dù cho đến nay ACOS chưa có được một định nghĩa phổ quát dựa trên nền tảng bệnh học nhưng đã có guideline đưa ACOS vào trong guideline COPD như là một phenotype (80). Trong một bài báo rất gần đây (năm 2017), Gustavo J Rodrigo và cs thậm trí còn nhấn mạnh rằng thuật ngữ ACOS không có ý nghĩa lâm sàng và nên bỏ (88a). Cho đến nay chúng ta cũng đã xem COPD như là một tình trạng rối loạn hơn là một bệnh. Tình trạng rối loạn đó là viêm mạn tính đường thở với biểu hiện tắc nghẽn cố định. Cho dù khởi đầu có yếu tố hút thuốc lá hay không thì ACOS vẫn có thể là giai đoạn cuối của hen phế quản và hen là yếu tố tăng nguy cơ COPD gấp 12 lần cao hơn những người không hen sau khi đã hiệu chỉnh yếu tố hút thuốc lá (88). GOLD 2017 (1) đã hoàn toàn có lý khi trích dẫn các nghiên cứu nhận định về kết cục COPD (tắc nghẽn cố định và giảm chỉ số khuếch tán) trên bệnh nhân hen. Tình trạng tắc nghẽn cố định trong ACOS luôn được nhấn mạnh với các đặc tính nhiều đợt cấp, đáp ứng tốt với corticosteroid và thông thường có tiên lượng xấu hơn. Như vậy, việc xem ACOS là COPD có kiểu hình hen (COPD asthmatic phenotype hay asthma-like phenotype) và có thể nằm trong nhóm exacerbator phenotype là dễ hiểu. Trên cơ sở này, chúng ta có thể chỉnh sửa lại sơ đồ phân loại COPD của GesEPOC thành dạng đơn giản, dễ hơn cho tiếp cận từ thực hành (hình 7).
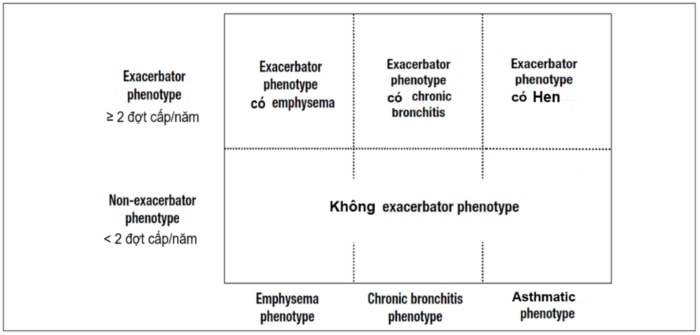
Hình 7. Phân loại phenotype lâm sàng VINACEEP 3 đề nghị dựa trên GesEPOC
Đợt cấp và phenotype:
Tình trạng triệu chứng của bệnh nhân có thể thay đổi trong một biên độ giao động nhất định thể hiện sự tương tác giữa tình trạng bệnh lý nền và tác động của yếu tố môi trường (bên ngoài hoặc bên trong) với phản ứng của cơ thể. Do yếu tố dự đoán đợt cấp COPD mạnh nhất là tiền sử đợt cấp nên đã đưa đến việc phynotype bệnh nhân thành kiểu hình nhiều đợt cấp và không nhiều đợt cấp như chúng ta đã bàn ở trên (mục 4). Thông thường tác nhân gây đợt cấp là virus, vi khuẩn, nhưng những tác nhân do nhiễm phải (acquisition) mới này với tình trạng viêm và colonization có mối quan hệ như thế nào vẫn còn là điều chúng ta hiểu chưa rõ (89). Điều này dẫn đến giả thuyết cho rằng sẽ có nhiều yếu tố tác động đến cơ chế hình thành đợt cấp, không giống nhau giữa người này và người khác và việc nghiên cứu các marker sinh học sẽ là một hứa hẹn. Trong tình trạng ổn định, những bệnh nhân có vi khuẩn phân lập được trong đàm sẽ có nguy cơ đợt cấp nhiễm khuẩn (OR 4,9), trong khi những bệnh nhân tăng BCAT trong đàm sẽ có nguy cơ đợt cấp tăng BCAT (OR 2,7) (90). Trong một nghiên cứu, Bafadhel M và cs (năm 2011) (50) ghi nhận 55%, 29% và 28% các trường hợp đợt cấp kết hợp với vi khuẩn, virus và tăng BACT đàm. Như vậy, việc phenotype đợt cấp có ý nghĩa quan trọng trong xử trí đợt cấp. Nó sẽ giúp cho thầy thuốc định hướng vai trò của corticosteroid và kháng sinh trong tình huống này. Mặc dù chưa có bằng chứng chắc chắn, tăng BCAT trong đàm có nhiều khả năng lên quan tới nhiễm virus và hiện tượng tăng BCAT trong đàm, máu cho thấy có khả năng đợt cấp có lợi khi điều trị bằng corticosteroid toàn thân (91). Vollenweider và cs trong một phân tích Cochrane (năm 2012) cho thấy lợi ích của trị liệu kháng sinh chỉ thấy rõ và ổn định trên nhóm bệnh nhân đợt cấp nặng nhập ICU (92). Đối với những bệnh nhân còn lại và điều trị ngoại trú, điều trị kháng sinh không làm thay đổi kết cục: tử vong, thất bại điều trị và ngày nằm viện có ý nghĩa. Các tác giả cũng cho rằng cần có những nghiên cứu kết hợp lâm sàng và biomarker để xác định nhóm bệnh nhân cần điều trị kháng sinh.
Trong đợt cấp, có sự gia tăng tình trạng viêm tại chỗ và toàn thân (93). Trong tình trạng này có sự gia tăng của BCĐNTT, lymphocyte, BCAT trong đường thở và đàm (94-96). Nhiều chất trung gian hóa học lưu hành tăng lên cả trong giai đoạn ổn định và trong đợt cấp. C-reactive protein là marker viêm hệ thống tăng trong đợt cấp và có vẻ như có tham gia vào dòng thác viêm (97). Với quan điểm tích hợp các mục tiêu xử trí đợt cấp với giảm gánh nặng hậu quả viêm trong COPD, việc điều trị có thể hướng tới làm giảm CRP, như là tác động vào dòng thác viêm (98).
Chẩn đoán COPD trong thực hành:
Thực hành chẩn đoán, kể cả chẩn đoán sớm, cần phải phải quyết định 2 vấn đề: xác định COPD và định hướng phenotype COPD. Tiếp cận và xử trí bệnh nhân COPD thông thường là công việc của các bác sỹ đa khoa và tuyến y tế ban đầu (99). Trong bối cảnh thực tế của Việt Nam, việc xây dựng một lộ trình tiếp cận (pathway) hợp lý với các yêu cầu khoa học, đơn giản và phân tuyến là rất cần thiết.
Về chẩn đoán: Cần lưu ý đến những người từ 40 trở lên có triệu chứng hô hấp kéo dài (kể cả bệnh nhân hen đang được điều trị kiểm soát), nhất là khi bệnh nhân có hút thuốc lá. Tất cả các tài liệu hướng dẫn, kể cả của Hội Lao và bệnh phổi Việt Nam, đều khuyến cáo cần xác định hội chứng thông khí tắc nghẽn với FEV1/FVC <0,7 để chẩn đoán COPD. Tuy nhiên cũng cần nhìn nhận rằng việc này mang nhiều ý nghĩa lý thuyết hơn là thực tiễn. Các lý do mang tính kỹ thuật trở ngại cho chẩn đoán COPD như sau: 1) Thông thường thầy thuốc thực hành ít nghĩ đến COPD khi bệnh nhân đến khám vì triệu chứng đợt cấp. Trong những tình huống ấy, bệnh nhân thường được chẩn đoán viêm phế quản hoặc hen (100). 2) Kiến thức và việc sử dụng kết quả đo chức năng hô hấp trong chẩn đoán COPD còn rất hạn chế (101). Thực hành COPD ở cộng đồng có thể đến 50% không đo chức năng hô hấp (102,103). Trong một nghiên cứu gần đây ở Tây Ban Nha, ghi nhận cả ở nông thôn và thành thị, số bệnh nhân chẩn đoán COPD được đo chỉ trên 20% (104). Một nhóm tác giả Việt Nam đã xây dựng bảng điểm chẩn đoán COPD (cho bệnh nhân từ 60 tuổi trở lên, có hút thuốc lá) mà trong đó chỉ cần khám và khai thác triệu chứng lâm sàng mạn tính và tiền sử khám, điều trị bệnh phổi đã có khả năng chẩn đoán COPD với diện tích dưới đường cong >0,9 (105). Khi có các triệu chứng bất thường nghi ngờ, không nằm trong bệnh cảnh phổ biến của COPD (thí dụ sụt cân, ho máu, sốt nhẹ dai dẳng, khò khè cơn, phù ngoại vi...), thì việc chụp Xquang ngực (hay CT ngực), đo chức năng phổi, siêu âm tim là cần thiết và là khả thi trong tình hình thực hành hiện nay ở Việt Nam.
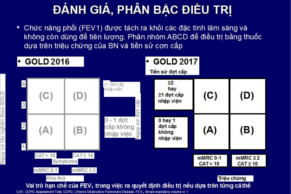
Về đánh giá phân loại mức độ nặng: Đánh giá mức độ nặng của tắc nghẽn dựa trên giá trị FEV1 cũng đã được khuyến cáo trên tất cả các tài liệu hướng dẫn. Việc sử dụng phân loại mức độ nặng của tình trạng tắc nghẽn để hướng dẫn điều trị cũng là một trở ngại lớn trong thực hành. Trong một nghiên cứu năm 2011 ở Mỹ, Gregory D Salinas và cs nhận thấy chỉ có 57,6 – 61,6% các bác sỹ nội khoa và bác sỹ gia đình sử dụng kết quả chức năng phổi để hướng dẫn điều trị (106). Cho đến nay phân loại mức độ nặng của tắc nghẽn cơ bản chỉ còn ở 2 mức (FEV1 ≥ 50% và <50% giá trị ước tính). Để giảm nhẹ các trở ngại cho thực hành chẩn đoán và đánh giá, Hội lao và bệnh phổi Việt nam (năm 2015) đã đề xuất phân tuyến, trong đó nhiệm vụ xác định chẩn đoán bằng chức năng phổi và Xquang ngực là nhiệm vụ của tuyến 2 (tuyến khám và điều trị có giường lưu và/hoặc có triển khai khám chuyên khoa hóa hô hấp) (hình 8)(107). Tuy nhiên việc này cũng chưa hẳn đã áp dụng dễ dàng vào được thực tế vì tính ràng buộc trách nhiệm giữa các tuyến ở Việt Nam còn mang nhiều ý nghĩa tượng trưng. Do vậy, một giải pháp lâm sàng cho việc đánh giá mức độ nặng của tình trạng tắc nghẽn là cần thiết và trên cơ sở giải pháp lâm sàng đó, giảm nhẹ yêu cầu đánh giá bằng mMRC và CAT như GOLD khuyến cáo vì thực chất 2 tiêu chuẩn đánh giá này cũng không tương quan (not identical) và cũng đã có ý kiến cho rằng chỉ nên sử dụng 1 trong hai tiêu chuẩn này (108).
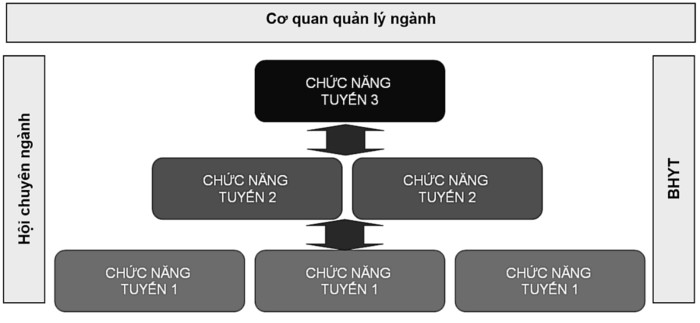
Hình 8. Mô hình hệ thống quản lý và điều trị Hen và COPD do Hội lao và bệnh phổi Việt Nam đề xuất
Một nghiên cứu nhận thấy nếu bệnh nhân có mMRC từ mức 3 điểm (khó thở khi đi bộ chậm trên mặt bằng trong vòng 100m hoặc sau vài phút) trở lên thì nhiều khả năng bệnh nhân đã ở mức độ tắc nghẽn trung bình-nặng (109). Trong một nghiên cứu khác, D. Dhanalakshmi và cs (năm 2016) (110) ghi nhận bệnh nhân ở GOLD III (tức là FEV1<50% giá trị ước tính) có mMRC là 2,73 ± 0,45 điểm. Như vậy, bằng khám lâm sàng và hỏi bệnh để có được một giá trị đánh giá mức độ nặng sử dụng cho khởi đầu việc quản lý và điều trị là khả thi. Một hệ thống đánh giá và quản lý bệnh nhân cần được thiết kế như khuyến cáo của Hội lao và bệnh phổi năm 2015 (107) và tích hợp từng trường hợp vào hệ thống này là cách tiếp cận hợp lý.
Về phenotype: Cho đến nay, như phân tích ở trên, chúng ta không thể chỉ bằng một marker nào đó để phenotype bệnh nhân trên một nền tảng bệnh học đa chiều như trong COPD. Như vậy, về thực chất, việc phenotype là xác định hai nhóm bệnh nhân: có lợi hay không khi kết hợp điều trị với ICS.
Dự đoán khả năng có lợi với điều trị kết hợp corticosteroid:
Với đặc tính không đồng nhất về bản chất bệnh học, các nghiên cứu với mẫu lớn do không phân tích subgroup nên đã không cho thấy nhóm bệnh nhân nào sẽ có lợi rõ khi điều trị với ICS (111,112). Nhiều ý kiến cho rằng rất cần nhưng cho đến nay các nghiên cứu theo phương pháp phân tích tổ hợp (cluster analysis) chưa xác định được một cách rõ ràng phenotype có lợi trong việc điều trị bằng corticosteroid (113-116). Dường như có mối liên hệ giữa 3 thành tố trong bệnh học COPD ở một nhóm bệnh nhân có lợi với điều trị corticosteroid: Bệnh lý đưởng thở nhỏ ưu thế - Tăng viêm, nhất là tăng bạch cầu ái toan và Nhiều đợt cấp (113).
Về bản chất bệnh học như trên đã phân tích, đặc tính viêm và tái tạo cấu trúc trong hen và COPD khác nhau nhưng cũng có sự chồng lấp, trong đó bao gồm viêm, tổn thương và tái tạo ở đường thở nhỏ, trong đó đường thở nhỏ là vị trí bệnh lý quan trọng. Bình thường đường thở nhỏ chỉ chiếm 10% trở kháng đường thở nhưng trong COPD, tổn thương đường thở nhỏ là cơ chế chủ yếu của tăng trở kháng (117) và có vai trò như là yếu tố quyết định tình trạng tắc nghẽn ở giai đoạn đầu (65,118). Trong đó 3 hình thái tổn thương quan trọng của viêm là tái tạo, thu hẹp và biến mất các tiểu phế quản tận. Trong một nghiên cứu trên COPD nặng và khí phế thũng dạng trung tâm tiểu thùy cho thấy số lượng các tiểu phế quản tận giảm xuống 10 lần và tổng diện tích cắt ngang giảm 100 lần (119). Một cách suy diễn hợp lý rằng các bất thường này là do quá trình viêm và thoái biến cấu trúc đã tạo ra (117). Như vậy việc ngăn chặn quá trình viêm là hướng tiếp cận điều trị cần thiết. Trong mục 4 có đề cập tới 2 nghiên cứu rất gần đây (67,68) cho thấy nếu ngưng ICS trên bệnh nhân COPD nặng-rất nặng có ít nhất 1 đợt cấp trong năm trước đó sẽ làm tăng tốc độ giảm FEV1 sau đó, trong khi nghiên cứu kia nhận thấy hiệu quả kháng viêm sẽ không còn nếu ngưng ICS. Tính chất viêm không bị khống chế khi ngưng ICS thể hiện trên cả tổng lượng tế bào và các loại tế bào gồm cả các lymphocyte, BCĐNTT và đại thực bào trên bệnh phẩm là đàm và mảnh sinh thiết phế quản (68). Điều này cho thấy các biến đổi trên đường thở, có thể không kết hợp chặt chẽ với đợt cấp, nhưng kết hợp quan trọng với thoái biến chức năng hô hấp. Từ đó cho thấy viêm và kháng viêm bằng ICS là quan trọng với tiêu chí làm giảm tốc độ thoái biến chức năng hô hấp, ít nhất là ở một phân nhóm bệnh nhân có tổn thương chủ yếu ở đường thở nhỏ.
Nhiều nghiên cứu đã cho thấy bệnh nhân COPD có tăng một số marker viêm trong máu, nhất là CRP (120,121). Tình trạng viêm toàn thân thông qua các marker viêm cho thấy hình ảnh không đồng nhất kiểu hình trong COPD không chỉ ở bản thân bệnh học COPD mà còn ở các biểu hiện toàn thân và bệnh đồng mắc (như xương khớp, tim mạch). Câu hỏi về việc liệu các marker viêm này có giúp hướng dẫn điều trị luôn được đặt ra trong nhiều nghiên cứu. CRP là marker dễ thực hiện, dễ đánh giá trong thực hành và cũng là marker được nghiên cứu nhiều nhất trong COPD (122). Mặc dù tăng CRP được xem là hậu quả của tình trạng viêm trong COPD (123) mà không phải là biểu hiện của nguyên nhân (117) nhưng nhiều nghiên cứu đã xác nhận vai trò tiên lượng của tăng CRP trong COPD như yếu tố dự đoán tăng tử vong, tăng đợt cấp, tăng nhập viện (120-125). Trong COPD, corticosteroid có thể làm giảm CRP ở tình trạng tiền viêm (proinflammatory state) (126,127) và làm giảm tình trạng tăng phản ứng đường thở liên quan tới COPD (128). Trong một nghiên cứu, Don D. Sin và cs (2004)(129) nhận thấy ngưng ICS thì ngưỡng CRP tăng và khi sử dụng trở lại CRP giảm có ý nghĩa. Tình trạng giảm CRP được duy trì ổn định trên 4 tháng trong giai đoạn sử dụng ICS. Các tác giả cho rằng ICS có thể làm giảm viêm toàn thân và điều này là có lợi trong COPD. Trong một phân tích gộp (meta analysis) Wen Qi Gan và cs (năm 2005)(130) nhận thấy ICS làm giảm tất cả các tế bào viêm trong đàm, trong đó có cả tế bào biểu mô, lymphocyte và BCĐNTT. Như vậy, ICS làm giảm viêm, hình ảnh viêm được phản ánh gián tiếp qua CRP và điều trị ICS làm giảm tế bào viêm và CRP (129,130).
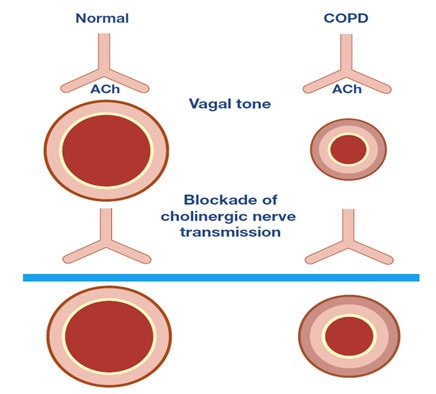
Vai trò anti-muscarinic receptor trong COPD:
Các muscarinic receptor (MR) có mặt trên hầu hết các tế bào ở phổi và tham gia điều hành các tín hiệu tế bào có bản chất acetylcholine (131). Ở phổi người, MR tập trung nhiều ở tế bào cơ trơn, tế bào biểu mô và các fibroblast. Thuốc kháng muscarinic được chỉ định rộng rãi trong COPD và một phần trong hen. Thoạt đầu, kháng muscarinic được chỉ định như là thuốc dãn phế quản, nhưng hiện nay nhóm thuốc này được xem là có kết hợp với hiệu quả kháng viêm, ức chế tăng sinh và kháng lại tình trạng tái tạo cấu trúc (131). Hiện tượng tái tạo cấu trúc trên đường thở nhỏ là đặc tính bệnh học chủ yếu trong COPD. Bên cạnh đó, các MR cũng có tác dụng điều phối tính hằng định nội môi glycosaminoglycans, nhất là hyaluronic acid và matrix metalloproteases là các phân tử có vai trò lớn trong viêm và biến đổi cấu trúc khung mô phổi trong COPD và hen. Đặc tính dược lý kháng viêm, kháng tái tạo cấu trúc trong COPD và hen của nhóm thuốc kháng muscarinic đang là đối tượng của các nghiên cứu lâm sàng.
Trong sinh bệnh học bệnh phổi tắc nghẽn như COPD và hen, có hiện tượng tăng kích thích hệ thống thần kinh phó giao cảm tạo ra co thắt cơ trơn và tăng tiết. Các thuốc kháng MR đã chứng minh được hiệu quả điều trị trong COPD và hen là do trương lực phó giao cảm có thể hồi phục tình trạng thu hẹp lòng đường thở (132). Ipratropium là thuốc được giới thiệu đầu tiên có khả năng ức chế các MR1,2,3 và sau đó là tiotropium bromide monohydrate có khả năng chọn lọc cao trên MR3 và duy trì thời gian tác dụng kéo dài. Với đặc tính dược lý như vậy, tiotropium cải thiện được tình trạng khó thở, khả năng gắng sức, giảm tình trạng căng phồng phổi quá mức (hyperinflation) và giảm đợt cấp trên bệnh nhân COPD trung bình-nặng (133). Hơn nữa, có bằng chứng trên súc vật thí nghiệm và người về hiện tượng khiếm khuyết trình diện và/hoặc kích thích MR ở phổi bệnh nhân COPD và hen. Chức năng tự ức chế của MR2 trên đường thở bệnh nhân không hoạt động bình thường và mất chức năng MR2 làm gia tăng phản ứng với kích thích (thí dụ như kháng nguyên trong hen) (134). Hiện tượng tái tạo đường thở có thể cũng kết hợp với tăng acetylcholine do khiếm khuyết chức năng MR2 (135). Trong hen nặng, khó trị và COPD có tăng phản ứng các nghiên cứu cho thấy có sự gia tăng có ý nghĩa MR3 (136,137). Điều này cho thấy có các cơ chế phân tử xảy ra không giống nhau tạo nên các hình thái co thắt phế quản trong hen nặng và COPD. Trong COPD và hen, MR còn hoạt động có liên quan tới acetylcholine từ thần kinh hoặc không thần kinh (neuronal, non-neuronal acetylcholine) (138-140).
Tình trạng tắc nghẽn đường thở trong COPD, như đã phân tích, là do co thắt cơ trơn, tăng tiết, viêm cùng với các biến đổi tái tạo trên đường thở và mất lực co hồi trun của nhu mô phổi. Trị liệu cần hướng tới đầu tiên trong COPD là cải thiện tắc nghẽn. So với kích thích beta2, kháng MR tỏ ra là hiệu quả hơn đối với các cơ chế tắc nghẽn kém hồi phục. Tác dụng dãn cơ trơn trong COPD của kháng MR tốt hơn kích thích beta2 trên người già (do giảm hiệu ứng beta2 receptor ở người già, có thể do giảm số lượng beta2 receptor) (141-143) và methylxanthine. Bên cạnh đó kháng MR còn ít kích thích trên tim hơn. Kết hợp kháng MR và kích thích beta2 tạo ra hiệu ứng hiệp đồng (synergy) do bổ sung tác dụng dược lý và thời gian tác dụng nên hiệu quả tốt hơn so với từng thuốc đơn độc, kể cả khi đã tăng liều (1).
6. Khuyến cáo thực hành:
Về chẩn đoán:
Trong hệ thống chăm sóc y tế hiện nay ở Việt Nam, việc áp dụng các cách tiếp cận phù hợp để chẩn đoán COPD, nhất là chẩn đoán sớm, một cách linh hoạt là rất cần thiết. Hiện nay, có nhiều khả năng các xét nghiệm máu, Xquang ngực thực hiện dễ dàng hơn thực hiện đo chức năng phổi. Các triệu chứng lâm sàng gợi ý COPD nên được xác định với một hình Xquang ngực không có hình mờ bất thường có thể lý giải các triệu chứng hô hấp mạn tính trên người bệnh. Do vậy việc nghĩ tới và chẩn đoán COPD lâm sàng (không cần xác định bằng giá trị FEV1/FVC) là có thể chấp nhận. Tuy nhiên, trong mọi trường hợp, khi điều trị theo hướng COPD tỏ ra không cải thiện, khi có những triệu chứng bất thường không nằm trong bệnh cảnh COPD thì cần chẩn đoán phân biệt và chuyển tuyến khi không có điều kiện và đủ kinh nghiệm. COPD là bệnh mạn tính nên khi đã có chẩn đoán COPD, nhất thiết bệnh nhân phải được đặt trong một quy trình theo dõi, quản lý điều trị sau đó. Ngay cả khi đó là tiếp xúc lần đầu với bệnh nhân trong bệnh viện vì đợt cấp.
Việc phân loại mức độ nặng và phân loại kiểu hình cần được thực hiện ngay trước khi quyết định điều trị với tối thiểu là phân nhóm bệnh nhân nhiều hay ít đợt cấp. Nếu có điều kiện thì thực hiện thêm xét nghiệm máu: công thức bạch cầu (xác định BCAT, tỷ lệ BCĐNTT/ lymphocyte) và CRP. Hình ảnh Xquang ngực có thể giúp nhận định về phenotype khí phế thũng hay phenotype phế quản. Phenotype trong COPD không phải là bất biến, việc đánh giá liên tục để điều chỉnh điều trị là cần thiết. Mục tiêu của điều trị là tình trạng lâm sàng ổn định và không để xảy ra đợt cấp.
Hình ảnh Xquang ngực, các xét nghiệm máu có thể giúp tăng thêm giá trị xác định phenotype có lợi trong việc điều trị bằng ICS kết hợp. Trong trường hợp không có điều kiện đo chức năng hô hấp, phân loại mức độ nặng có thể đơn giản hóa bằng các câu hỏi xác định mức độ khó thở gắng sức tương đương với mMRC 2-3 điểm.
Nên xác định một cơ sở chuyên khoa (chức năng tuyến 2 hoặc 3) có khả năng xác định sâu hơn về phenotype để hỗ trợ chẩn đoán và có hướng dẫn điều trị thích hợp khi điều trị ban đầu không hiệu quả.
Về điều trị thuốc:
Trong COPD, khác với hen, trừ khi bệnh nhân chỉ khó thở không thường xuyên (intermittent), nên khởi đầu điều trị ngay với thuốc dãn phế quản tác dụng kéo dài, ngay cả ở giai đoạn nhẹ, tốt hơn sử dụng thuốc dãn phế quản tác dụng ngắn. Ở tình trạng ổn định, nếu năng lực gắng sức tương đương mMRC ≥ 2-3 điểm nên khởi đầu điều trị ngay bằng thuốc dãn phế quản tác dụng kéo dài kết hợp cho phân nhóm ít đợt cấp bằng LAMA + LABA và bằng ICS-LABA + LAMA cho phân nhóm nhiều đợt cấp.
Nên cân nhắc điều trị thêm theophylline dạng phóng thích chậm (SR) cho cả hai nhóm nếu trị liệu như trên tỏ ra kém kiểm soát, nhất là khi có nhiều đợt cấp. Liều duy trì 2-7mg/kg/ngày.
Đối với nhóm tổn thương không ưu thế khí phế thũng, nếu trị liệu ban đầu tỏ ra kém hiệu quả, nhất là khi có nhiều đợt cấp, nên cân nhắc kết hợp điều trị kéo dài thêm thuốc tan đàm (mucolytic), macrolide.
Cần xác định tuyến cho cơ sở khám và điều trị của mình. Cần xây dựng mối quan hệ chặt chẽ về chuyên môn trong hệ thống. Chẩn đoán xác định COPD có tính khách quan, phân nhóm phenotype, đánh giá khi không đáp ứng điều trị, chẩn đoán phân biệt, thực hiện các xét nghiệm chuyên biệt nên được triển khai đầy đủ ở tuyến 3 (tuyến chuyên khoa cuối trong đơn vị tỉnh).
Kết luận:
Bệnh phổi tắc nghẽn mạn tính nên được nhìn nhận và tiếp cận điều trị từ bản chất viêm. Với tình hình thực tế ở Việt Nam như hiện nay, chẩn đoán sớm COPD và chẩn đoán định hướng bản chất viêm là khả thi. Tiếp cận điều trị và điều trị sớm theo bản chất viêm là hướng đi hứa hẹn hiệu quả điều trị cao, giảm chi phí điều trị và giảm tác dụng ngoại ý do sử dụng thuốc quá mức. Xây dựng lộ trình tiếp cận thực hành trong COPD có tính quy ước là quan trọng và các nghiên cứu đánh giá cần được thực hiện để có được những khuyến cáo có giá trị khoa học cao trong thời gian tới.
Dưới đây là sơ đồ hướng dẫn chẩn đoán phân loại và điều trị thuốc trong COPD có cân nhắc thực tế thuốc lưu hành phổ biến ở Việt Nam.
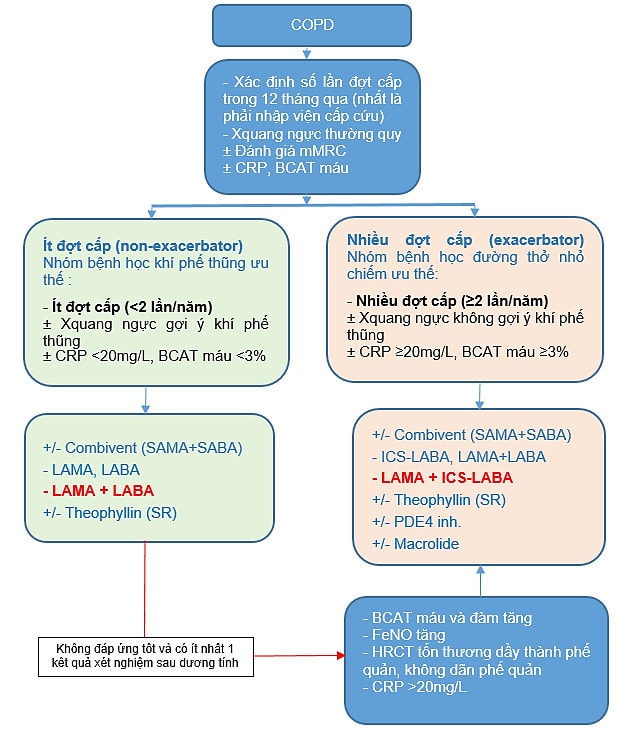
Ý nghĩa các dấu: “±”: Có hay không; “+/-“: Cân nhắc để kết hợp hay không; “…-…”: Thuốc dạng kết hợp; “+”: Kết hợp thuốc; “,” : Hoặc; Chữ mầu đỏ: Khởi đầu điều trị nếu mRC >2-3 điểm. Mũi tên đỏ: Chuyển tuyến
Hình 9. Sơ đồ tiếp cận điều trị COPD Hội Lao và Bệnh phổi Việt Nam đề nghị
Tài liệu tham khảo:
|
1. |
Global Initiative for Chronic Obstructive Lung Disease (report 2017) |
|
2. |
B. L. Barker and C. E. Brightling. Phenotyping the heterogeneity of chronic obstructive pulmonary disease. Clinical Science (2013) 124, 371–387 |
|
3. |
Roberts, S. D., Farber, M. O., Knox, K. S., Phillips, G. S., Bhatt, N. Y., Mastronarde, J. G. And Wood, K. L. (2006) FEV1/FVC ratio of 70% misclassifies patients with obstruction at the extremes of age. Chest 130, 200–206 |
|
4. |
Hardie, J. A., Buist, A. S., Vollmer, W. M., Ellingsen, I., Bakke, P. S. and Morkve, O. (2002) Risk of over-diagnosis of COPD in asymptomatic elderly never-smokers. Eur. Respir. J. 20, 1117–1122 |
|
5. |
Han, M. K., Agusti, A., Calverley, P. M., Celli, B. R., Criner, G., Curtis, J. L., Fabbri, L. M., Goldin, J. G., Jones, P. W., Macnee, W. Et al. (2010) Chronic obstructive pulmonary disease phenotypes: the future of COPD. Am. J. Respir. Crit. Care Med. 182, 598–604 |
|
6. |
Nicolas Roche, Céline Pribil, Eric Van Ganse et al. Real-life use of fluticasone propionate/salmeterol in patients with chronic obstructive pulmonary disease: a French observational studyBMC Pulmonary Medicine 2014, 14:56 |
|
7. |
Claudio Tantucci, Laura Pini. COPD: it is time to change!. International Journal of COPD 2015:10 2451–2457 |
|
8. |
K.F. Chung and I.M. Adcock. Multifaceted mechanisms in COPD: inflammation, immunity, and tissue repair and destruction. Eur Respir J 2008; 31: 1334–1356 |
|
9. |
Turino, G. M., Seniorrm, Garg, B. D., Keller, S., Levi, M. M. And Mandl, I. (1969) Serum elastase inhibitor deficiency and α1-antitrypsin deficiency in patients with obstructive emphysema. Science 165, 709–711 |
|
10. |
Imai, K., Mercer, B. A., Schulman, L. L., Sonett, J. R. And D’Armiento, J. M. (2005) Correlation of lung surface area to apoptosis and proliferation in human emphysema. Eur. Respir. J. 25, 250–258 |
|
11. |
Yokohori, N., Aoshiba, K. And Nagai, A. (2004) Increased levels of cell death and proliferation in alveolar wall cells in patients with pulmonary emphysema. Chest 125, 626–632 |
|
12. |
Hawgood, S., Ochs, M., Jung, A., Akiyama, J., Allen, L., Brown, C., Edmondson, J., Levitt, S., Carlson, E. And Gillespie, A. M. (2002) Sequential targeted deficiency of SP-A and –D leads to progressive alveolar lipoproteinosis and emphysema. Am. J. Physiol. Lung Cell. Mol. Physiol. 283, L1002 – L1010 |
|
13. |
Nagai, A., West, W. W. And Thurlbeck, W. M. (1985) The National Institutes of Health intermittent positive-pressure breathing trial: Pathology studies. II. Correlation between morphologic findings, clinical findings, and evidence of expiratory air-flow obstruction. Am. Rev. Respir. Dis. 132, 946–953 |
|
14. |
Saetta, M., Turato, G., Maestrelli, P., Mapp, C. E. And Fabbri, L. M. (2001) Cellular and structural bases of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 163, 1304–1309 |
|
15. |
Maestrelli, P., Saetta, M., Mapp, C. E. And Fabbri, L. M. (2001) Remodeling in response to infection and injury. Airway inflammation and hypersecretion of mucus in smoking subjects with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 164, S76–80 |
|
16. |
Hogg, J. C., Chu, F., Utokaparch, S., Woods, R., Elliott, W. M, Buzatu, L., Cherniack, R. M., Rogers, R. M., Sciurba, F. C., Coxson, H. O. And Par´e, P. D. (2004) The nature of small-airway obstruction in chronic obstructive pulmonary disease. N. Engl. J. Med. 350, 2645–2653 |
|
17. |
Hogg, J. C. (2004) Pathophysiology of airflow limitation in chronic obstructive pulmonary disease. Lancet 364, 709–721 |
|
18. |
McDonough, J. E., Yuan, R., Suzuki, M., Seyednejad, N., Elliott, W. M., Sanchez, P. G., Wright, A. C., Gefter, W. B., Litzky, L., Coxson, H. O. Et al. (2011) Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N. Engl. J. Med. 365, 1567–1575 |
|
19. |
Foster, W. M., Langenback, E. G. And Bergofsky, E. H. (1985) Disassociation in the mucociliary function of central and peripheral airways of asymptomatic smokers. Am. Rev. Respir. Dis. 132, 633–639 |
|
20. |
Raman, A. S., Swinburne, A. J. And Fedullo, A. J. (1983) Pneumococcal adherence to the buccal epithelial cells of cigarette smokers. Chest 83, 23–27 |
|
21. |
Herr, C., Beisswenger, C., Hess, C., Kandler, K., Suttorp, N., Welte, T., Schroeder, J. M. And Vogelmeier, C. (2009) Suppression of pulmonary innate host defence in smokers. Thorax 64, 144–149 |
|
22. |
Polosukhin, V. V., Cates, J. M., Lawson, W. E., Zaynagetdinov, R., Milstone, A. P., Massion, P. P., Ocak, S., Ware, L. B., Lee, J. W., Bowler, R. P. Et al. (2011) Bronchial secretory immunoglobulin a deficiency correlates with airway inflammation and progression of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 184, 317–327 |
|
23. |
Hodge, S., Hodge, G., Ahern, J., Jersmann, H., Holmes, M. And Reynolds, P. N. (2007) Smoking alters alveolar macrophage recognition and phagocytic ability: Implications in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 37, 748–755 |
|
24. |
Verra, F., Escudier, E., Lebargy, F., Bernaudin, J. F., De Cremoux, H. And Bignon, J. (1995) Ciliary abnormalities in bronchial epithelium of smokers, ex-smokers, and nonsmokers. Am. J. Respir. Crit. Care Med. 151, 630–634 |
|
25. |
Charles S. Berenson, Mary Alice Garlipp, Lori J. Grove, Jane Maloney, Sanjay Sethi. Impaired Phagocytosis of Nontypeable Haemophilus influenzae by Human Alveolar Macrophages in Chronic Obstructive Pulmonary Disease. The Journal of Infectious Diseases 2006; 194:1375–84 |
|
26. |
Taylor, A. E., Finney-Hayward, T. K., Quint, J. K., Thomas, C. M., Tudhope, S. J., Wedzicha, J. A., Barnes, P. J. And Donnelly, L. E. (2010) Defective macrophage phagocytosis of bacteria in COPD. Eur. Respir. J. 35, 1039–1047 |
|
27. |
Bethan L. Barker, Christopher E. Brightling. Phenotyping the heterogeneity of chronic obstructive pulmonary diseaseClinical Science (2013) 124, 371–387 |
|
28. |
Polosukhin, V. V., Cates, J. M., Lawson, W. E., Zaynagetdinov, R., Milstone, A. P., Massion, P. P., Ocak, S., Ware, L. B., Lee, J. W., Bowler, R. P. Et al. (2011) Bronchial secretory immunoglobulin a deficiency correlates with airway inflammation and progression of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 184, 317–327 |
|
29. |
M.G. Cosio Piqueras, M.G. Cosio. Disease of the airways in chronic obstructive pulmonary disease. Eur Respir J 2001; 18: Suppl. 34, 41s–49s |
|
30. |
Dirkje S. Postma, Scott T. Weiss, Maarten van den Berge, et al. Revisiting the Dutch hypothesis. J Allergy Clin Immunol 2015;136:521-9 |
|
31. |
Tashkin DP, Altose MD, Bleecker ER, Connett JE, Kanner RE, Lee WW, et al. The lung health study: airway responsiveness to inhaled methacholine in smokers with mild to moderate airflow limitation. The Lung Health Study Research Group. Am Rev Respir Dis 1992;145:301-10. |
|
32. |
Van den Berge M, Vonk JM, Gosman M, Lapperre TS, Snoeck-Stroband JB, Sterk PJ, et al. Clinical and inflammatory determinants of bronchial hyperresponsiveness in COPD. Eur Respir J 2012;40:1098-105. |
|
33. |
Postma DS, Wempe JB, Renkema TE, Van Der Mark TW, Koeter GH. Hyperresponsiveness as a determinant of the outcome in chronic obstructive pulmonary disease. Am Rev Respir Dis 1991;143:1458-62. |
|
34. |
Diana C. Grootendorst, Klaus F. Rabe. Mechanisms of Bronchial Hyperreactivity in Asthma and Chronic Obstructive Pulmonary Disease. Proc Am Thorac Soc Vol 1. Pp 77–87, 2004 |
|
35. |
Nicola Scichilone, Salvatore Battaglia, Alba La Sala, Vincenzo Bellia. Clinical implications of airway hyperresponsiveness in COPD. International Journal of COPD 2006:1(1) 49–60 |
|
36. |
O’Byrne PM, Postma DS. The many faces of airway inflammation. Asthma and chronic obstructive pulmonary disease. Asthma Research Group. Am J Respir Crit Care Med 1999;159:S41-63. |
|
37. |
Brightling CE. Eosinophils, bronchitis and asthma: pathogenesis of cough and airflow obstruction. Pulm Pharmacol Ther 2011;24:324-7. |
|
38. |
Van den Berge M, Meijer RJ, Kerstjens HA, de Reus DM, Koeter GH, Kauffman HF, et al. PC(20) adenosine 59-monophosphate is more closely associated with airway inflammation in asthma than PC(20) methacholine. Am J Respir Crit Care Med 2001;163:1546-50. |
|
39. |
Broekema M, Timens W, Vonk JM, Volbeda F, Lodewijk ME, Hylkema MN, et al. Persisting remodeling and less airway wall eosinophil activation in complete remission of asthma. Am J Respir Crit Care Med 2011;183:310-6. |
|
40. |
Brightling CE, McKenna S, Hargadon B, Birring S, Green R, Siva R, et al. Sputum eosinophilia and the short term response to inhaled mometasone in chronic obstructive pulmonary disease. Thorax 2005;60:193-8. |
|
41. |
Siva R, Green RH, Brightling CE, Shelley M, Hargadon B, McKenna S, et al. Eosinophilic airway inflammation and exacerbations of COPD: a randomised controlled trial. Eur Respir J 2007;29:906-13. |
|
42. |
Bathoorn E, Liesker JJ, Postma DS, Koeter GH, van der Toorn M, van der Heide S, et al. Change in inflammation in out-patient COPD patients from stable phase to a subsequent exacerbation. Int J Chron Obstruct Pulmon Dis 2009;4:101-9. |
|
43. |
Dave Singh, Umme Kolsum, Chris E. Brightling, Nicholas Locantore, Alvar Agusti and Ruth Tal-Singer on behalf of the ECLIPSE investigators. Eosinophilic inflammation in COPD: prevalence and clinical characteristics Eur Respir J 2014; 44: 1697–1700 |
|
44. |
Liesker JJ, Bathoorn E, Postma DS, Vonk JM, Timens W, Kerstjens HA. Sputum inflammation predicts exacerbations after cessation of inhaled corticosteroids in COPD. Respir Med 2011;105:1853-60. |
|
45. |
Ghebre MA, Bafadhel M, Desai D, Cohen SE, Newbold P, Rapley L, et al. Biological clustering supports both ‘‘Dutch’’ and ‘‘British’’ hypotheses of asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol 2015;135:63-72. |
|
46. |
Brightling CE, McKenna S, Hargadon B, et al. Sputum eosinophilia and the short term response to inhaled mometasone in chronic obstructive pulmonary disease. Thorax 2005; 60: 193–198. |
|
47. |
Brightling CE, Monteiro W, Ward R, et al. Sputum eosinophilia and short-term response to prednisolone in chronic obstructive pulmonary disease: a randomised controlled trial. Lancet 2000; 356: 1480–1485. |
|
48. |
Leigh R, Pizzichini MM, Morris MM, et al. Stable COPD: predicting benefit from high-dose inhaled corticosteroid treatment. Eur Respir J 2006; 27: 964–971. |
|
49. |
Pizzichini E, Pizzichini MM, Gibson P, et al. Sputum eosinophilia predicts benefit from prednisone in smokers with chronic obstructive bronchitis. Am J Respir Crit Care Med 1998; 158: 1511–1517. |
|
50. |
Bafadhel M, McKenna S, Terry S, et al. Acute exacerbations of chronic obstructive pulmonary disease: identification of biologic clusters and their biomarkers. Am J Respir Crit Care Med 2011; 184: 662–671. |
|
51. |
Siva R, Green RH, Brightling CE, et al. Eosinophilic airway inflammation and exacerbations of COPD: a randomised controlled trial. Eur Respir J 2007; 29: 906–913. |
|
52. |
Bafadhel M, McKenna S, Terry S, et al. Blood eosinophils to direct corticosteroid treatment of exacerbations of chronic obstructive pulmonary disease: a randomized placebo-controlled trial. Am J Respir Crit Care Med 2012; 186: 48–55. |
|
53. |
R. Siva, R.H. Green, C.E. Brightling, M. Shelley, B. Hargadon, S. McKenna, W. Monteiro, M. Berry, D. Parker, A.J. Wardlaw and I.D. Pavord. Eosinophilic airway inflammation and exacerbations of COPD: a randomised controlled trialEur Respir J 2007; 29: 906–913 |
|
54. |
Mona Bafadhel, Susan McKenna, Sarah Terry, et al. Blood Eosinophils to Direct Corticosteroid Treatment of Exacerbations of Chronic Obstructive Pulmonary Disease A Randomized Placebo-Controlled Trial. Am J Respir Crit Care Med Vol 186, Iss. 1, pp 48–55, Jul 1, 2012 |
|
55. |
Cosio MG, Saetta M, Agusti A. Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med 2009; 360: 2445–54. |
|
56. |
Nunez B, Sauleda J, Anto JM, et al. Anti-tissue antibodies are related to lung function in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2011; 183: 1025–31. |
|
57. |
Greene CM, Low TB, O’Neill SJ, McElvaney NG. Anti-proline-glycine-proline or antielastin autoantibodies are not evident in chronic infl ammatory lung disease. Am J Respir Crit Care Med 2010; 181: 31–35. |
|
58. |
Lee SH, Goswami S, Grudo A, et al. Antielastin autoimmunity in tobacco smoking-induced emphysema. Nat Med 2007; 13: 567–69. |
|
59. |
M. Saetta, W.D. Kim, J.L. Izquierdo, H. Ghezzo et al. Extent of centrilobular and panacinar emphysema in smokers’ lungs: pathological and mechanical implicationsEur Respir J, 1994, 7, 664–671 |
|
60. |
J. Hogg. Peripheral lung remodelling in asthma and chronic obstructive pulmonary disease. Eur Respir J 2004; 24: 893–894 |
|
61. |
Turato G, Zuin R, Miniati M, Baraldo S, Rea F, Beghe B, et al: Airway inflammation in severe chronic obstructive pulmonary disease: relationship with lung function and radiologic emphysema. Am J Respir Crit Care Med 2002; 166: 105–110. |
|
62. |
Retamales I, Elliott WM, Meshi B, Coxson HO, Pare PD, Sciurba FC, et al: Amplification of inflammation in emphysema and its association with latent adenoviral infection. Am J Respir Crit Care Med 2001; 164: 469– 473. |
|
63. |
Nagai A, West WW, Paul JL, Thurlbeck WM: The National Institutes of Health Intermittent Positive Pressure Breathing trial: pathology studies. I. Interrelationship between morphologic lesions. Am Rev Respir Dis 1985; 132: 937–945. |
|
64. |
John E. McDonough, Ren Yuan, Masaru Suzuki. Small-Airway Obstruction and Emphysema in Chronic Obstructive Pulmonary Disease N Engl J Med 365;17 nejm.org october 27, 2011 |
|
65. |
Seemungal TA et al. Effect of exacerbation on quality of life in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;157:1418-22 |
|
66. |
John R. Hurst,Jorgen Vestbo, Antonio Anzueto et al. Susceptibility to Exacerbation in Chronic Obstructive Pulmonary Disease. N Engl J Med 2010;363:1128-38. |
|
67. |
Helgo Magnussen, Bernd Disse, Roberto Rodriguez-Roisin et al. Withdrawal of Inhaled Glucocorticoids and Exacerbations of COPD. N Engl J Med 2014;371:1285-94. |
|
68. |
Lisette I.Z. Kunz, Nick H.T. ten Hacken, Thérèse S. Lapperre et al. Airway inflammation in COPD after long-term withdrawal of inhaled corticosteroids. Eur Respir J 2017; 49: 1600839 |
|
69. |
Anthony D’Urzo, James F Donohue, Peter Kardos et al. A re-evaluation of the role of inhaled corticosteroids in the management of patients with chronic obstructive pulmonary disease. Expert Opin. Pharmacother. (2015) 16(12):1845-1860 |
|
70. |
Haughney J, Gruffydd-Jones K, Roberts J, et al. The distribution of COPD in UK general practice using the new GOLD classification. Eur Respir J 2014;43:993-1002 |
|
71. |
Martinez-Garcia MA´ , Soler-Catalun~a JJ, Donat SY, et al. Factors associated with bronchiectasis in patients with COPD. Chest 2011;140:1130-7 |
|
72. |
Patel IS, Vlahos I, Wilkinson TM, et al. Bronchiectasis, exacerbation indices, and inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2004;170:400-7 |
|
73. |
Jennifer A Dickens, Bruce E Miller, Lisa D Edwards et al. COPD association and repeatability of blood biomarkers in the ECLIPSE cohort. Respiratory Research 2011, 12:146 |
|
74. |
Agustí A et al. Persistent systemic inflammation is associated with poor clinical outcomes in COPD: a novel phenotype. PloS One. 2012;7:e37483 |
|
75. |
Thomsen M et al. Inflammatory biomarkers and exacerbations in chronic obstructive pulmonary disease. JAMA. 2013;309:2353-61. |
|
76. |
Aida M. Yousef, Wael Alkhiary. Role of neutrophil to lymphocyte ratio in prediction of acute exacerbation of chronic obstructive pulmonary disease. Egypt. J. Chest Dis. Tuberc. (2016), http://dx.doi.org/10.1016/j.ejcdt.2016.09.006. |
|
77. |
C. E. Brightling. Biomarkers that Predict and Guide Therapy for Exacerbations of Chronic Obstructive Pulmonary Disease. Ann Am Thorac Soc Vol 10, Supplement, pp S214–S219, Dec 2013 |
|
78. |
Brendan J. Carolan, E. Rand Sutherland. Clinical phenotypes of chronic obstructive pulmonary disease and asthma: Recent advances. J Allergy Clin Immunol 2013;131:627-34 |
|
79. |
Claudio Tantucci, Laura Pini. COPD: it is time to change!. International Journal of COPD 2015:10 2451–2457 |
|
80. |
Miravitlles M, Soler-Cataluna JJ, Calle M, et al. Spanish guideline for COPD (GesEPOC). Update 2014. Arch Bronconeumol 2014;50(Suppl 1):1-16 |
|
81. |
Global Initiative for Asthma. Global Strategy for Asthma Management and Prevention, 2016. Available from: www.ginasthma.org |
|
82. |
Antonis Papaiwannou, Paul Zarogoulidis, Konstantinos Porpodis et al. Asthma-chronic obstructive pulmonary disease overlap syndrome (ACOS): current literature review. J Thorac Dis 2014;6(S1):S146-S151. |
|
83. |
Chalmers GW, Macleod KJ, Little SA, et al. Influence of cigarette smoking on inhaled corticosteroid treatment in mild asthma. Thorax 2002;57:226-30. |
|
84. |
Chaudhuri R, Livingston E, McMahon AD, et al. Cigarette smoking impairs the therapeutic response to oral corticosteroids in chronic asthma. Am J Respir Crit Care Med 2003;168:1308-11. |
|
85. |
Chanez P, Vignola AM, O’Shaugnessy T, et al. Corticosteroid reversibility in COPD is related to features of asthma. Am J Respir Crit Care Med 1997;155:1529-34 |
|
86. |
Hamid Q, Tulic MK. New insights into the pathophysiology of the small airways in asthma. Ann Thorac Med 2007;2:28-33. |
|
87. |
Park JW, Hong YK, Kim CW, et al. High resolution computed tomography in patients with bronchial asthma: correlation with clinical features, pulmonary function and bronchial hyperresponsiveness. J Investig Allergol Clin Immunol 1997;7:186-92. |
|
88. |
Bosken CH, Wiggs BR, Pare PD, et al. Small airway dimensions in smokers with obstruction to airflow. Am Rev Respir Dis 1990;142:563-70. |
|
88a |
Gustavo J Rodrigo, Hugo Neffen, Vicente Plaza. ACOS: a controversial concept. Curr Opin Allergy Clin Immunol 217. 17: 36-41. |
|
89. |
Menezes AM, de Oca MM, Perez-Padilla R, et al. Increased risk of exacerbation and hospitalization in subjects with an overlap phenotype: COPD-asthma. Chest 2014;145:297-304. |
|
90. |
Graciela E. Silva, Duane L. Sherrill, Stefano Guerra et al. Asthma as a Risk Factor for COPD in a Longitudinal Study. Chest 2004; 126:59–65 |
|
91. |
C. E. Brightling. Biomarkers that Predict and Guide Therapy for Exacerbations of Chronic Obstructive Pulmonary Disease. Ann Am Thorac Soc Vol 10, Supplement, pp S214–S219, Dec 2013 |
|
92. |
Vollenweider DJ, Jarrett H, Steurer-Stey CA, Garcia-Aymerich J, Puhan MA. Antibiotics for exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2012;12: CD010257. |
|
93. |
Walters JA, Gibson PG, Wood-Baker R, Hannay M, Walters EH. Systemic corticosteroids for acute exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2009;(1):CD001288. |
|
94. |
Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. New England Journal of Medicine 2008; 359 (22):2355–65. |
|
95. |
Papi A, Bellettato CM, Braccioni F, Romagnoli M, Casolari P, Caramori G, et al. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. American Journal of Respiratory and Critical Care Medicine 2006; 173 (10):1114–21. |
|
96. |
Bathoorn E, Kerstjens H, Postma D, Timens W, MacNee W. Airways inflammation and treatment during acute exacerbations of COPD. International Journal of Chronic Obstructive Pulmonary Disease 2008; 3 (2):217–29. |
|
97. |
Falk JA, Minai OA, Mosenifar Z. Inhaled and systemic corticosteroids in chronic obstructive pulmonary disease. Proceedings of the American Thoracic Society; 2008 May 16-20; Toronto. 2008. |
|
98. |
E.F. Wouters, K.H. Groenewegen, M.A. Dentener, J.H. Vernooy, Systemic inflammation in chronic obstructive pulmonary disease: the role of exacerbations, Proc. Am. Thorac. Soc. 4 (8) (2007) 626–634. |
|
99. |
Barr RG, Celli BR, Martinez FJ, et al. Physician and patient perceptions in COPD: the COPD Resource Network Needs Assessment Survey. Am J Med. 2005;118(12):1415 |
|
100. |
Tinkelman DG, Price DB, Nordyke RJ, Halbert RJ. Misdiagnosis of COPD and asthma in primary care patients 40 years of age and over. J Asthma. 2006; 43(1):75–80. |
|
101. |
Kaminsky DA, Marcy TW, Bachand M, Irvin CG. Knowledge and use of office spirometry for the detection of chronic obstructive pulmonary disease by primary care physicians. Respir Care. 2005;50(12): 1639–1648. |
|
102. |
Bolton CE, Ionescu AA, Edwards PH, Faulkner TA, Edwards SM, Shale DJ. Attaining a correct diagnosis of COPD in general practice. Respir Med 2005;99:493–500. |
|
103. |
Joo MJ, Lee TA, Weiss KB. Geographic variation of spirometry use in newly diagnosed COPD. Chest 2008;134:38–45. |
|
104.. |
Eduardo Márquez-Martín, Joan B Soriano, Myriam Calle Rubio et al. Differences in the use of spirometry between rural and urban primary care centers in Spain. International Journal of COPD 2015:10 1633–1639 |
|
105. |
Nguyễn Văn Thành, Cao Thị Mỹ Thúy, Võ Phạm Minh Thư và cs. Xây dựng mô hình hệ thống quản lý và điều trị hiệu quả COPD và Hen phế quản trong bệnh viện và ở cộng đồng. NXB Y Học 2012, tr. 57-69 |
|
106. |
Gregory D Salinas, James C Williamson, Ravi Kalhan et al. Barriers to adherence to chronic obstructive pulmonary disease guidelines by primary care physicians. International Journal of COPD 2011:6 171–179 |
|
107. |
Hội lao và Bệnh phổi Việt nam. Hướng dẫn quốc gia xử trí hen và bệnh phổi tắc nghẽn mạn tính. Nhà xuất bản y học 2015. Tr. 13-15 |
|
108. |
Sunmin Kim, Jisun Oh, Yu-Il Kim et al. Differences in classification of COPD group using COPD assessment test (CAT) or modified Medical Research Council (mMRC) dyspnea scores: a cross-sectional analyses. BMC Pulmonary Medicine 2013, 13:35 |
|
109. |
Kylie Hill PhD Richard Hodder et al. Identifying adults at risk of COPD who need confirmatory spirometry in primary care. Vol 57: february 2011;57:51-7 |
|
110. |
D. Dhanalakshmi, Sai Savya, Sravan kumar et al. Relationship between Dyspnoea MMRC Scale and Forced Expiratory Volume in First Second (FEV1) In Chronic Obstructive Pulmonary Disease. Sch. J. App. Med. Sci., 2016; 4(9E):3544-3547 |
|
111. |
R. Leigh, M.M.M. Pizzichini, M.M. Morris. Stable COPD: predicting benefit from high-dose inhaled corticosteroid treatment. Eur Respir J 2006; 27: 964–971 |
|
112. |
Burge PS. EUROSCOP, ISOLDE and the Copenhagen city lung study. Thorax 1999; 54: 287–288. |
|
113. |
Weatherall M, Travers J, Shirtcliffe PM, et al. Distinct clinical phenotypes of airways disease defined by cluster analysis. Eur Respir J 2009; 34: 812–818. |
|
114. |
Burgel PR, Paillasseur JL, Caillaud D, et al. Clinical COPD phenotypes: a novel approach using principal component and cluster analyses. Eur Respir J 2010; 36: 531–539. |
|
115. |
Garcia-Aymerich J, Gomez FP, Benet M, et al. Identification and prospective validation of clinically relevant chronic obstructive pulmonary disease (COPD) subtypes. Thorax 2011; 66: 430–437. |
|
116. |
J. Travers, M. Weatherall, J. Fingleton et al. Towards individualised medicine for airways disease: identifying clinical phenotype groups. Eur Respir J 2012; 39: 1033–1048 |
|
117. |
Hogg JC, Macklem PT, Thurlbeck WM. Site and nature of airway obstruction in chronic obstructive lung disease. N Engl J Med 1968; 278: 1355–1360. |
|
118. |
P-R. Burgel. The role of small airways in obstructive airway diseases. Eur Respir Rev 2011; 20: 119, 23–33 |
|
119. |
Hogg JC, McDonough JE, Sanchez PG, et al. Micro-computed tomography measurements of peripheral lung pathology in chronic obstructive pulmonary disease. Proc Am Thorac Soc 2009; 6: 546–549. |
|
120. |
Pinto-Plata VM, Mullerova H, Toso JF, et al. C-reactive protein in patients with COPD, control smokers and non-smokers. Thorax 2006; 61: 23–28. |
|
121. |
Yanbaeva DG, Dentener MA, Spruit MA, et al. IL6 and CRP haplotypes are associated with COPD risk and systemic inflammation: a case-control study. BMC Med Genet 2009; 10: 23 |
|
122 |
Emer Kelly, Caroline A Owen, Victor Pinto-Plata et al. The Role of Systemic Inflammatory Biomarkers to Predict Mortality in Chronic Obstructive Pulmonary Disease. Expert Rev Resp Med. 2013;7(1):57-64. |
|
123. |
Morten Dahl, Jørgen Vestbo, Peter Lange et al. C-reactive Protein As a Predictor of Prognosis in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med Vol 175. pp 250–255, 2007 |
|
124. |
Morten Dahl, Jørgen Vestbo, Jeppe Zacho, et al. C reactive protein and chronic obstructive pulmonary disease: a Mendelian randomisation approach. Thorax 2011;66:197-204. |
|
125. |
T.M.L. Eagan, T. Ueland, P.D. Wagner et al. Systemic inflammatory markers in COPD: results from the Bergen COPD Cohort Study. Eur Respir J 2010; 35: 540–548 |
|
126. |
Azar RR, Rinfret S, Theroux P, Stone PH, Dakshinamurthy R, Feng YJ,Wu AH, Range G, Waters DD. A randomized placebo-controlled trial to assess the efficacy of antiinflammatory therapy with methylprednisolone in unstable angina (MUNA trial). Eur Heart J 2000;21: 2026–2032. |
|
127. |
Versaci F, Gaspardone A, Tomai F, Ribichini F, Russo P, Proietti I, Ghini AS, Ferrero V, Chiariello L, Gioffre PA, et al. Immunosuppressive Therapy for the Prevention of Restenosis after Coronary Artery Stent Implantation (IMPRESS Study). J Am Coll Cardiol 2002;40:1935–1942. |
|
128. |
The Lung Health Study Research Group. Effect of inhaled triamcinolone on the decline in pulmonary function in chronic obstructive pulmonary disease. N Engl J Med 2000;343:1902–1909. |
|
129. |
Don D. Sin, Paige Lacy, Ernest York et al. Effects of Fluticasone on Systemic Markers of Inflammation in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med Vol 170. pp 760–765, 2004 |
|
130. |
Wen Qi Gan, SF Paul Man, Don D Sin. Effects of inhaled corticosteroids on sputum cell counts in stable chronic obstructive pulmonary disease: a systematic review and a meta-analysis. BMC Pulmonary Medicine 2005, 5:3 |
|
131. |
George Karakiulakis, Michael Roth. Muscarinic Receptors and Their Antagonists in COPD: Anti-Inflammatory and Antiremodeling Effects. Mediators of Inflammation Volume 2012, Article ID 409580, 9 pages |
|
132. |
R. Gosens, J. Zaagsma, H. Meurs, A. J. Halayko. Muscarinic receptor signaling in the pathophysiology of asthma and COPD. Respiratory Research, vol. 7, article 73, 2006. |
|
133. |
J. A. Ohar and J. F. Donohue, Mono- and combination therapy of long-acting bronchodilators and inhaled corticosteroids in advanced COPD. Seminars in Respiratory and Critical Care Medicine, vol. 31, no. 3, pp. 321–333, 2010. |
|
134. |
Z. Nie, D. B. Jacoby, A. D. Fryer. Etanercept prevents airway hyperresponsiveness by protecting neuronal M2 muscarinic receptors in antigen-challenged guinea pigs. British Journal of Pharmacology, vol. 156, no. 1, pp. 201–210, 2009. |
|
145. |
S. Matthiesen, A. Bahulayan, S. Kempkens et al. Muscarinic receptors mediate stimulation of human lung fibroblast proliferation. American Journal of Respiratory Cell and Molecular Biology, vol. 35, no. 6, pp. 621–627, 2006. |
|
136. |
P. A. Selivanova, E. S. Kulikov,O. V. Kozina, E. A. Gereng, M. B. Freidin, and L.M. Ogorodova. Morphological and molecular characteristics of “difficult” asthma. Journal of Asthma, vol. 47, no. 3, pp. 269–275, 2010. |
|
137. |
P. A. Selivanova, E. S. Kulikov, O. V. Kozina et al. Differential expression of the β,2-adrenoreceptor and M3-cholinoreceptor genes in bronchial mucosa of patients with asthma and chronic obstructive pulmonary disease. Annals of Allergy, Asthma & Immunology, vol. 108, no. 1, pp. 39–43, 2012. |
|
138. |
L. E. Kistemaker, T. A. Oenema, H. Meurs, and R. Gosens. Regulation of airway inflammation and remodeling by muscarinic receptors: perspectives on anticholinergic therapy in asthma and COPD. Life Sciences, vol. 91, no. 21-22, pp. 1126–1133, 2012. |
|
139. |
R. Gosens, I. S. T. Bos, J. Zaagsma, and H. Meurs, Protective effects of tiotropium bromide in the progression of airway smooth muscle remodeling. American Journal of Respiratory and Critical Care Medicine, vol. 171, no. 10, pp. 1096–1102, 2005. |
|
140. |
C. L. Grainge, L. C. K. Lau, J. A. Ward et al. Effect of bronchoconstriction on airway remodeling in asthma, The New England Journal of Medicine, vol. 364, no. 21, pp. 2006–2015, 2011. |
|
141. |
van Schayck CP, Folgering H, Harbers H, et al. Effects of allergy and age on responses to salbutamol and ipratropium bromide in moderate asthma and chronic bronchitis. Thorax 1991;46:355–9. |
|
142. |
Ullah MI, Newman GB, Saunders KB. Influence of age in response to ipratropium and salbutamol in asthma. Thorax 1981;36:523–9. |
|
143. |
Appleton S, Jones T, Poole P, Lasserson TJ et al. Ipratropiumbromide versus long-acting beta-2 agonists for stable chronic obstructive pulmonary disease (Review). Cochrane Database of Systematic Reviews 2006, Issue 3. Art. No.: CD006101. |